Decision of the Optimal Rank of a Nonnegative Matrix Factorization Model for Gene Expression Data Sets Utilizing the Unit Invariant Knee Method: Development and Evaluation of the Elbow Method for Rank Selection.
{"title":"Decision of the Optimal Rank of a Nonnegative Matrix Factorization Model for Gene Expression Data Sets Utilizing the Unit Invariant Knee Method: Development and Evaluation of the Elbow Method for Rank Selection.","authors":"Emine Guven","doi":"10.2196/43665","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>There is a great need to develop a computational approach to analyze and exploit the information contained in gene expression data. The recent utilization of nonnegative matrix factorization (NMF) in computational biology has demonstrated the capability to derive essential details from a high amount of data in particular gene expression microarrays. A common problem in NMF is finding the proper number rank (r) of factors of the degraded demonstration, but no agreement exists on which technique is most appropriate to utilize for this purpose. Thus, various techniques have been suggested to select the optimal value of rank factorization (r).</p><p><strong>Objective: </strong>In this work, a new metric for rank selection is proposed based on the elbow method, which was methodically compared against the cophenetic metric.</p><p><strong>Methods: </strong>To decide the optimum number rank (r), this study focused on the unit invariant knee (UIK) method of the NMF on gene expression data sets. Since the UIK method requires an extremum distance estimator that is eventually employed for inflection and identification of a knee point, the proposed method finds the first inflection point of the curvature of the residual sum of squares of the proposed algorithms using the UIK method on gene expression data sets as a target matrix.</p><p><strong>Results: </strong>Computation was conducted for the UIK task using gene expression data of acute lymphoblastic leukemia and acute myeloid leukemia samples. Consequently, the distinct results of NMF were subjected to comparison on different algorithms. The proposed UIK method is easy to perform, fast, free of a priori rank value input, and does not require initial parameters that significantly influence the model's functionality.</p><p><strong>Conclusions: </strong>This study demonstrates that the elbow method provides a credible prediction for both gene expression data and for precisely estimating simulated mutational processes data with known dimensions. The proposed UIK method is faster than conventional methods, including metrics utilizing the consensus matrix as a criterion for rank selection, while achieving significantly better computational efficiency without visual inspection on the curvatives. Finally, the suggested rank tuning method based on the elbow method for gene expression data is arguably theoretically superior to the cophenetic measure.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":" ","pages":"e43665"},"PeriodicalIF":0.0000,"publicationDate":"2023-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11135234/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/43665","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: There is a great need to develop a computational approach to analyze and exploit the information contained in gene expression data. The recent utilization of nonnegative matrix factorization (NMF) in computational biology has demonstrated the capability to derive essential details from a high amount of data in particular gene expression microarrays. A common problem in NMF is finding the proper number rank (r) of factors of the degraded demonstration, but no agreement exists on which technique is most appropriate to utilize for this purpose. Thus, various techniques have been suggested to select the optimal value of rank factorization (r).
Objective: In this work, a new metric for rank selection is proposed based on the elbow method, which was methodically compared against the cophenetic metric.
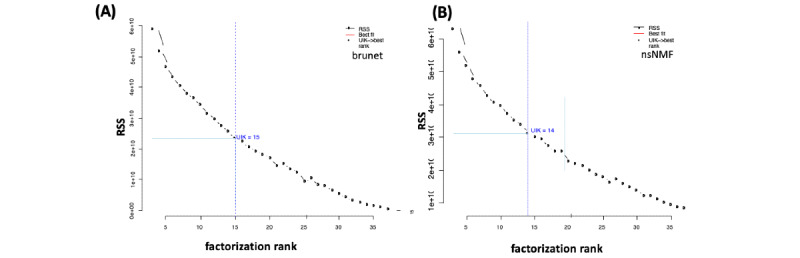
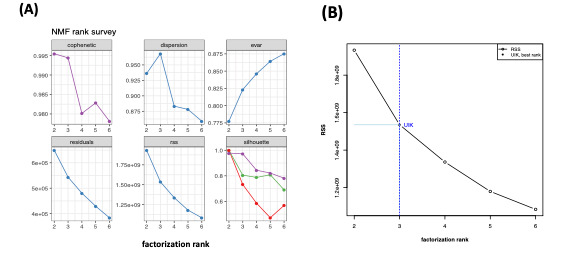
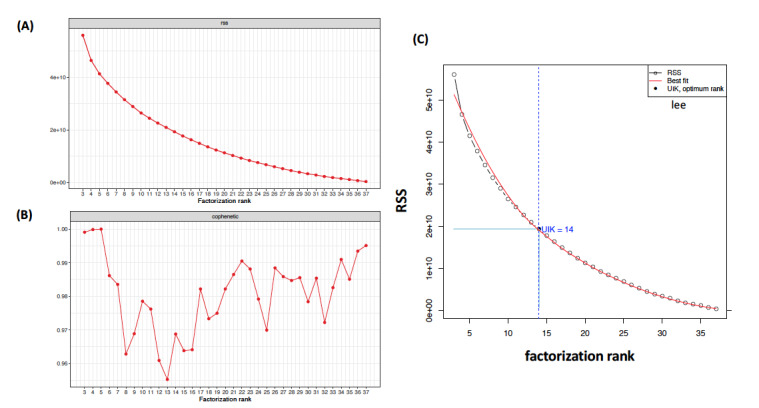
Methods: To decide the optimum number rank (r), this study focused on the unit invariant knee (UIK) method of the NMF on gene expression data sets. Since the UIK method requires an extremum distance estimator that is eventually employed for inflection and identification of a knee point, the proposed method finds the first inflection point of the curvature of the residual sum of squares of the proposed algorithms using the UIK method on gene expression data sets as a target matrix.
Results: Computation was conducted for the UIK task using gene expression data of acute lymphoblastic leukemia and acute myeloid leukemia samples. Consequently, the distinct results of NMF were subjected to comparison on different algorithms. The proposed UIK method is easy to perform, fast, free of a priori rank value input, and does not require initial parameters that significantly influence the model's functionality.
Conclusions: This study demonstrates that the elbow method provides a credible prediction for both gene expression data and for precisely estimating simulated mutational processes data with known dimensions. The proposed UIK method is faster than conventional methods, including metrics utilizing the consensus matrix as a criterion for rank selection, while achieving significantly better computational efficiency without visual inspection on the curvatives. Finally, the suggested rank tuning method based on the elbow method for gene expression data is arguably theoretically superior to the cophenetic measure.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
