Automated reaction kinetics and network exploration (Arkane): A statistical mechanics, thermodynamics, transition state theory, and master equation software
Alon Grinberg Dana, Matthew S. Johnson, Joshua W. Allen, Sandeep Sharma, Sumathy Raman, Mengjie Liu, Connie W. Gao, Colin A. Grambow, Mark J. Goldman, Duminda S. Ranasinghe, Ryan J. Gillis, A. Mark Payne, Yi-Pei Li, Xiaorui Dong, Kevin A. Spiekermann, Haoyang Wu, Enoch E. Dames, Zachary J. Buras, Nick M. Vandewiele, Nathan W. Yee, Shamel S. Merchant, Beat Buesser, Caleb A. Class, Franklin Goldsmith, Richard H. West, William H. Green
{"title":"Automated reaction kinetics and network exploration (Arkane): A statistical mechanics, thermodynamics, transition state theory, and master equation software","authors":"Alon Grinberg Dana, Matthew S. Johnson, Joshua W. Allen, Sandeep Sharma, Sumathy Raman, Mengjie Liu, Connie W. Gao, Colin A. Grambow, Mark J. Goldman, Duminda S. Ranasinghe, Ryan J. Gillis, A. Mark Payne, Yi-Pei Li, Xiaorui Dong, Kevin A. Spiekermann, Haoyang Wu, Enoch E. Dames, Zachary J. Buras, Nick M. Vandewiele, Nathan W. Yee, Shamel S. Merchant, Beat Buesser, Caleb A. Class, Franklin Goldsmith, Richard H. West, William H. Green","doi":"10.1002/kin.21637","DOIUrl":null,"url":null,"abstract":"<p>The open-source statistical mechanics software described here, Arkane–Automated Reaction Kinetics and Network Exploration–facilitates computations of thermodynamic properties of chemical species, high-pressure limit reaction rate coefficients, and pressure-dependent rate coefficient over multi-well molecular potential energy surfaces (PES) including the effects of collisional energy transfer on phenomenological kinetics. Arkane can use estimates to fill in information for molecules or reactions where quantum chemistry information is missing. The software solves the internal energy master equation for complex unimolecular reaction systems. Inputs to the software include converged electronic structure computations performed by the user using a variety of supported software packages (Gaussian, Molpro, Orca, TeraChem, Q-Chem, Psi4). The software outputs high-pressure limit rate coefficients and pressure-dependent phenomenological rate coefficients, as well as computed thermodynamic properties (enthalpy, entropy, and constant pressure heat capacity) with added energy corrections. Some of the key features of Arkane include treatment of 1D, 2D or ND hindered internal rotation modes, treatment of free internal rotation modes, quantum tunneling effect consideration, transition state theory (TST) and Rice-Ramsperger-Kassel-Marcus (RRKM) rate coefficient computations, master equation solution with four implemented methods, inverse-Laplace transform of high-pressure limit rate coefficients into the energy domain, energy corrections based on bond-additivity or isodesmic reactions, automated and efficient PES exploration, and PES sensitivity analysis. The present work describes the design of Arkane, how it should be used, and refers to the theory that it employs. Arkane is distributed via the RMG-Py software suite (https://github.com/ReactionMechanismGenerator/RMG-Py).</p>","PeriodicalId":13894,"journal":{"name":"International Journal of Chemical Kinetics","volume":"55 6","pages":"300-323"},"PeriodicalIF":1.6000,"publicationDate":"2023-04-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21637","citationCount":"9","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Chemical Kinetics","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/kin.21637","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 9
Abstract
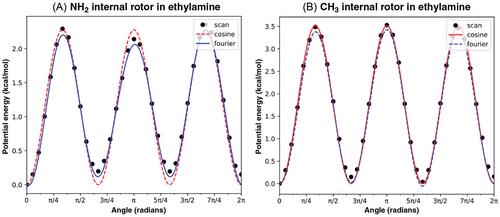
The open-source statistical mechanics software described here, Arkane–Automated Reaction Kinetics and Network Exploration–facilitates computations of thermodynamic properties of chemical species, high-pressure limit reaction rate coefficients, and pressure-dependent rate coefficient over multi-well molecular potential energy surfaces (PES) including the effects of collisional energy transfer on phenomenological kinetics. Arkane can use estimates to fill in information for molecules or reactions where quantum chemistry information is missing. The software solves the internal energy master equation for complex unimolecular reaction systems. Inputs to the software include converged electronic structure computations performed by the user using a variety of supported software packages (Gaussian, Molpro, Orca, TeraChem, Q-Chem, Psi4). The software outputs high-pressure limit rate coefficients and pressure-dependent phenomenological rate coefficients, as well as computed thermodynamic properties (enthalpy, entropy, and constant pressure heat capacity) with added energy corrections. Some of the key features of Arkane include treatment of 1D, 2D or ND hindered internal rotation modes, treatment of free internal rotation modes, quantum tunneling effect consideration, transition state theory (TST) and Rice-Ramsperger-Kassel-Marcus (RRKM) rate coefficient computations, master equation solution with four implemented methods, inverse-Laplace transform of high-pressure limit rate coefficients into the energy domain, energy corrections based on bond-additivity or isodesmic reactions, automated and efficient PES exploration, and PES sensitivity analysis. The present work describes the design of Arkane, how it should be used, and refers to the theory that it employs. Arkane is distributed via the RMG-Py software suite (https://github.com/ReactionMechanismGenerator/RMG-Py).
期刊介绍:
As the leading archival journal devoted exclusively to chemical kinetics, the International Journal of Chemical Kinetics publishes original research in gas phase, condensed phase, and polymer reaction kinetics, as well as biochemical and surface kinetics. The Journal seeks to be the primary archive for careful experimental measurements of reaction kinetics, in both simple and complex systems. The Journal also presents new developments in applied theoretical kinetics and publishes large kinetic models, and the algorithms and estimates used in these models. These include methods for handling the large reaction networks important in biochemistry, catalysis, and free radical chemistry. In addition, the Journal explores such topics as the quantitative relationships between molecular structure and chemical reactivity, organic/inorganic chemistry and reaction mechanisms, and the reactive chemistry at interfaces.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
