A Computational Study of the Electronic Energy and Charge Transfer Rates and Pathways in the Tetraphenyldibenzoperiflanthene/Fullerene Interfacial Dyad
Alexander Schubert, Srijana Bhandari, Eitan Geva* and Barry D. Dunietz*,
{"title":"A Computational Study of the Electronic Energy and Charge Transfer Rates and Pathways in the Tetraphenyldibenzoperiflanthene/Fullerene Interfacial Dyad","authors":"Alexander Schubert, Srijana Bhandari, Eitan Geva* and Barry D. Dunietz*, ","doi":"10.1021/acs.jpclett.3c01927","DOIUrl":null,"url":null,"abstract":"<p >The electronic transition rates and pathways underlying interfacial charge separation in tetraphenyldibenzoperiflanthene:fullerene (DBP:C<sub>70</sub>) blends are investigated computationally. The analysis is based on a polarization-consistent framework employing screened range-separated hybrid functional in a polarizable continuum model to parametrize Fermi’s golden rule rate theory. The model considers the possible transitions within the 25 lowest excited states of a DBP:C<sub>70</sub> dyad that are accessible by photoexcitation. The different identified pathways contributing to charge carrier generation include electron and hole transfer and backtransfer, exciton transfer, and internal relaxation steps. The larger density of states of C<sub>70</sub> appears to explain the previously observed larger efficiency for charge separation through hole transfer mechanism. We also analyze the validity of the high-temperature and short-time semiclassical approximations of the FGR theory, where both overestimated and underestimated Marcus theory based constants can be affected.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"14 43","pages":"9569–9583"},"PeriodicalIF":4.6000,"publicationDate":"2023-10-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01927","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
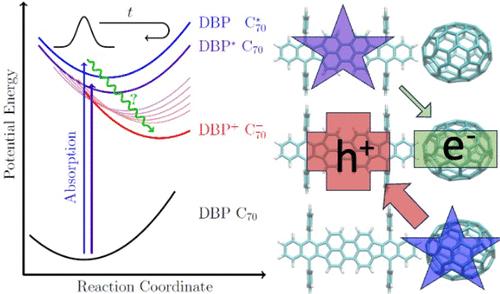
The electronic transition rates and pathways underlying interfacial charge separation in tetraphenyldibenzoperiflanthene:fullerene (DBP:C70) blends are investigated computationally. The analysis is based on a polarization-consistent framework employing screened range-separated hybrid functional in a polarizable continuum model to parametrize Fermi’s golden rule rate theory. The model considers the possible transitions within the 25 lowest excited states of a DBP:C70 dyad that are accessible by photoexcitation. The different identified pathways contributing to charge carrier generation include electron and hole transfer and backtransfer, exciton transfer, and internal relaxation steps. The larger density of states of C70 appears to explain the previously observed larger efficiency for charge separation through hole transfer mechanism. We also analyze the validity of the high-temperature and short-time semiclassical approximations of the FGR theory, where both overestimated and underestimated Marcus theory based constants can be affected.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
