Yi Fan, Changsu Cao, Xusheng Xu, Zhenyu Li, Dingshun Lv* and Man-Hong Yung*,
{"title":"Circuit-Depth Reduction of Unitary-Coupled-Cluster Ansatz by Energy Sorting","authors":"Yi Fan, Changsu Cao, Xusheng Xu, Zhenyu Li, Dingshun Lv* and Man-Hong Yung*, ","doi":"10.1021/acs.jpclett.3c01804","DOIUrl":null,"url":null,"abstract":"<p >Quantum computation represents a revolutionary approach to solving problems in quantum chemistry. However, due to the limited quantum resources in the current noisy intermediate-scale quantum (NISQ) devices, quantum algorithms for large chemical systems remain a major task. In this work, we demonstrate that the circuit depth of the unitary coupled cluster (UCC) and UCC-based ansatzes in the algorithm of the variational quantum eigensolver can be significantly reduced by an energy-sorting strategy. Specifically, subsets of excitation operators are first prescreened from the operator pool according to its contribution to the total energy. The quantum circuit ansatz is then iteratively constructed until convergence of the final energy to a typical accuracy. For demonstration, this method has been successfully applied to molecular and periodic systems. Particularly, a reduction of 50%–98% in the number of operators is observed while retaining the accuracy of the original UCCSD operator pools. This method can be straightforwardly extended to general parametric variational ansatzes.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"14 43","pages":"9596–9603"},"PeriodicalIF":4.6000,"publicationDate":"2023-10-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"14","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01804","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 14
Abstract
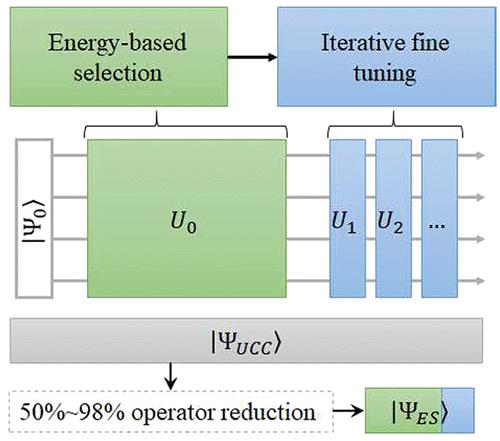
Quantum computation represents a revolutionary approach to solving problems in quantum chemistry. However, due to the limited quantum resources in the current noisy intermediate-scale quantum (NISQ) devices, quantum algorithms for large chemical systems remain a major task. In this work, we demonstrate that the circuit depth of the unitary coupled cluster (UCC) and UCC-based ansatzes in the algorithm of the variational quantum eigensolver can be significantly reduced by an energy-sorting strategy. Specifically, subsets of excitation operators are first prescreened from the operator pool according to its contribution to the total energy. The quantum circuit ansatz is then iteratively constructed until convergence of the final energy to a typical accuracy. For demonstration, this method has been successfully applied to molecular and periodic systems. Particularly, a reduction of 50%–98% in the number of operators is observed while retaining the accuracy of the original UCCSD operator pools. This method can be straightforwardly extended to general parametric variational ansatzes.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
