Di Zhu, Yiqiang Zhao, Ran Zhang, Hanyu Wu, Gengyuan Cai, Zhenfang Wu, Yuzhe Wang, Xiaoxiang Hu
{"title":"Genomic prediction based on selective linkage disequilibrium pruning of low-coverage whole-genome sequence variants in a pure Duroc population.","authors":"Di Zhu, Yiqiang Zhao, Ran Zhang, Hanyu Wu, Gengyuan Cai, Zhenfang Wu, Yuzhe Wang, Xiaoxiang Hu","doi":"10.1186/s12711-023-00843-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Although the accumulation of whole-genome sequencing (WGS) data has accelerated the identification of mutations underlying complex traits, its impact on the accuracy of genomic predictions is limited. Reliable genotyping data and pre-selected beneficial loci can be used to improve prediction accuracy. Previously, we reported a low-coverage sequencing genotyping method that yielded 11.3 million highly accurate single-nucleotide polymorphisms (SNPs) in pigs. Here, we introduce a method termed selective linkage disequilibrium pruning (SLDP), which refines the set of SNPs that show a large gain during prediction of complex traits using whole-genome SNP data.</p><p><strong>Results: </strong>We used the SLDP method to identify and select markers among millions of SNPs based on genome-wide association study (GWAS) prior information. We evaluated the performance of SLDP with respect to three real traits and six simulated traits with varying genetic architectures using two representative models (genomic best linear unbiased prediction and BayesR) on samples from 3579 Duroc boars. SLDP was determined by testing 180 combinations of two core parameters (GWAS P-value thresholds and linkage disequilibrium r<sup>2</sup>). The parameters for each trait were optimized in the training population by five fold cross-validation and then tested in the validation population. Similar to previous GWAS prior-based methods, the performance of SLDP was mainly affected by the genetic architecture of the traits analyzed. Specifically, SLDP performed better for traits controlled by major quantitative trait loci (QTL) or a small number of quantitative trait nucleotides (QTN). Compared with two commercial SNP chips, genotyping-by-sequencing data, and an unselected whole-genome SNP panel, the SLDP strategy led to significant improvements in prediction accuracy, which ranged from 0.84 to 3.22% for real traits controlled by major or moderate QTL and from 1.23 to 11.47% for simulated traits controlled by a small number of QTN.</p><p><strong>Conclusions: </strong>The SLDP marker selection method can be incorporated into mainstream prediction models to yield accuracy improvements for traits with a relatively simple genetic architecture, however, it has no significant advantage for traits not controlled by major QTL. The main factors that affect its performance are the genetic architecture of traits and the reliability of GWAS prior information. Our findings can facilitate the application of WGS-based genomic selection.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"72"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10583454/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00843-w","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Although the accumulation of whole-genome sequencing (WGS) data has accelerated the identification of mutations underlying complex traits, its impact on the accuracy of genomic predictions is limited. Reliable genotyping data and pre-selected beneficial loci can be used to improve prediction accuracy. Previously, we reported a low-coverage sequencing genotyping method that yielded 11.3 million highly accurate single-nucleotide polymorphisms (SNPs) in pigs. Here, we introduce a method termed selective linkage disequilibrium pruning (SLDP), which refines the set of SNPs that show a large gain during prediction of complex traits using whole-genome SNP data.
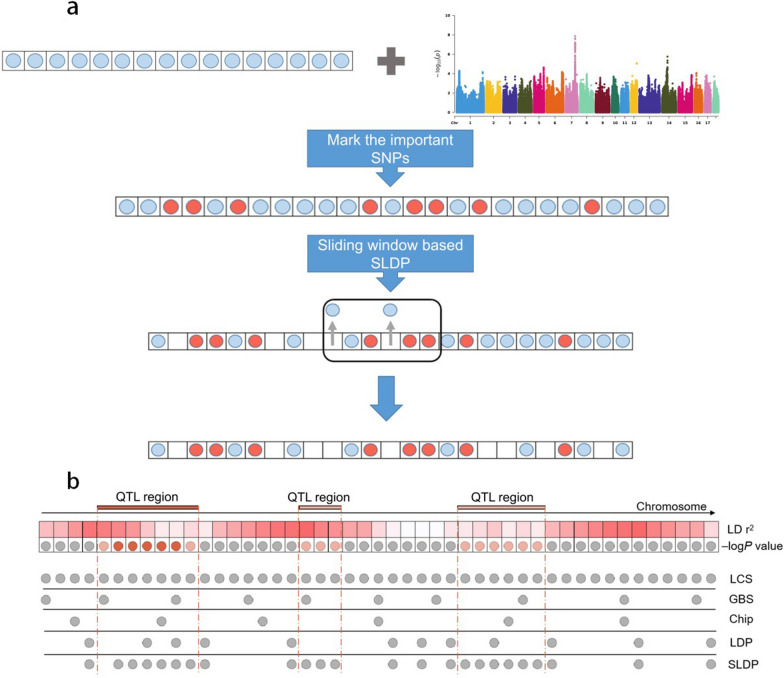
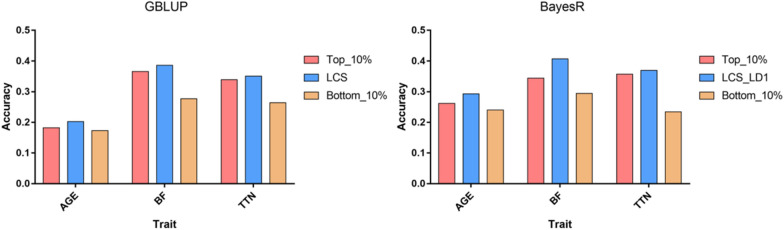
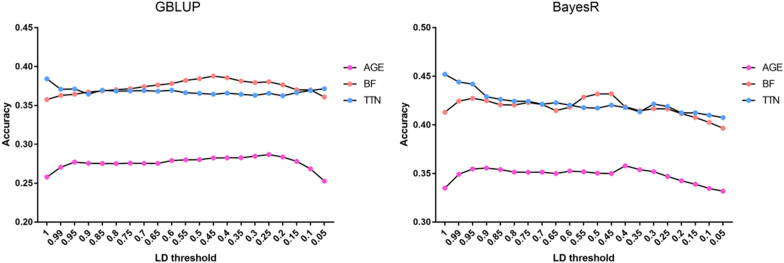
Results: We used the SLDP method to identify and select markers among millions of SNPs based on genome-wide association study (GWAS) prior information. We evaluated the performance of SLDP with respect to three real traits and six simulated traits with varying genetic architectures using two representative models (genomic best linear unbiased prediction and BayesR) on samples from 3579 Duroc boars. SLDP was determined by testing 180 combinations of two core parameters (GWAS P-value thresholds and linkage disequilibrium r2). The parameters for each trait were optimized in the training population by five fold cross-validation and then tested in the validation population. Similar to previous GWAS prior-based methods, the performance of SLDP was mainly affected by the genetic architecture of the traits analyzed. Specifically, SLDP performed better for traits controlled by major quantitative trait loci (QTL) or a small number of quantitative trait nucleotides (QTN). Compared with two commercial SNP chips, genotyping-by-sequencing data, and an unselected whole-genome SNP panel, the SLDP strategy led to significant improvements in prediction accuracy, which ranged from 0.84 to 3.22% for real traits controlled by major or moderate QTL and from 1.23 to 11.47% for simulated traits controlled by a small number of QTN.
Conclusions: The SLDP marker selection method can be incorporated into mainstream prediction models to yield accuracy improvements for traits with a relatively simple genetic architecture, however, it has no significant advantage for traits not controlled by major QTL. The main factors that affect its performance are the genetic architecture of traits and the reliability of GWAS prior information. Our findings can facilitate the application of WGS-based genomic selection.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
