{"title":"Accurate three-body noncovalent interactions: the insights from energy decomposition†","authors":"Sharon A. Ochieng and Konrad Patkowski","doi":"10.1039/D3CP03938B","DOIUrl":null,"url":null,"abstract":"<p >An impressive collection of accurate two-body interaction energies for small complexes has been assembled into benchmark databases and used to improve the performance of multiple density functional, semiempirical, and machine learning methods. Similar benchmark data on nonadditive three-body energies in molecular trimers are comparatively scarce, and the existing ones are practically limited to homotrimers. In this work, we present a benchmark dataset of 20 equilibrium noncovalent interaction energies for a small but diverse selection of 10 heteromolecular trimers. The new 3BHET dataset presents complexes that combine different interactions including π–π, anion–π, cation–π, and various motifs of hydrogen and halogen bonding in each trimer. A detailed symmetry-adapted perturbation theory (SAPT)-based energy decomposition of the two- and three-body interaction energies shows that 3BHET consists of electrostatics- and dispersion-dominated complexes. The nonadditive three-body contribution is dominated by induction, but its influence on the overall bonding type in the complex (as exemplified by its position on the ternary diagram) is quite small. We also tested the extended SAPT (XSAPT) approach which is capable of including some nonadditive interactions in clusters of any size. The resulting three-body dispersion term (obtained from the many-body dispersion formalism) is mostly in good agreement with the supermolecular CCSD(T)–MP2 values and the nonadditive induction term is similar to the three-body SAPT(DFT) data, but the overall three-body XSAPT energies are not very accurate as they are missing the first-order exchange terms.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 42","pages":" 28621-28637"},"PeriodicalIF":2.9000,"publicationDate":"2023-10-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03938b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
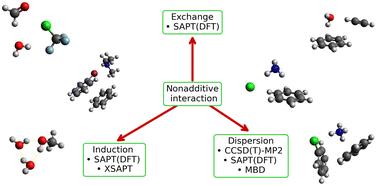
An impressive collection of accurate two-body interaction energies for small complexes has been assembled into benchmark databases and used to improve the performance of multiple density functional, semiempirical, and machine learning methods. Similar benchmark data on nonadditive three-body energies in molecular trimers are comparatively scarce, and the existing ones are practically limited to homotrimers. In this work, we present a benchmark dataset of 20 equilibrium noncovalent interaction energies for a small but diverse selection of 10 heteromolecular trimers. The new 3BHET dataset presents complexes that combine different interactions including π–π, anion–π, cation–π, and various motifs of hydrogen and halogen bonding in each trimer. A detailed symmetry-adapted perturbation theory (SAPT)-based energy decomposition of the two- and three-body interaction energies shows that 3BHET consists of electrostatics- and dispersion-dominated complexes. The nonadditive three-body contribution is dominated by induction, but its influence on the overall bonding type in the complex (as exemplified by its position on the ternary diagram) is quite small. We also tested the extended SAPT (XSAPT) approach which is capable of including some nonadditive interactions in clusters of any size. The resulting three-body dispersion term (obtained from the many-body dispersion formalism) is mostly in good agreement with the supermolecular CCSD(T)–MP2 values and the nonadditive induction term is similar to the three-body SAPT(DFT) data, but the overall three-body XSAPT energies are not very accurate as they are missing the first-order exchange terms.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
