{"title":"Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics.","authors":"Ireshyn Selvan Govender, Rethabile Mokoena, Stoyan Stoychev, Previn Naicker","doi":"10.3390/proteomes11040029","DOIUrl":null,"url":null,"abstract":"<p><p>Urine provides a diverse source of information related to a patient's health status and is ideal for clinical proteomics due to its ease of collection. To date, most methods for the preparation of urine samples lack the throughput required to analyze large clinical cohorts. To this end, we developed a novel workflow, urine-HILIC (uHLC), based on an on-bead protein capture, clean-up, and digestion without the need for bottleneck processing steps such as protein precipitation or centrifugation. The workflow was applied to an acute kidney injury (AKI) pilot study. Urine from clinical samples and a pooled sample was subjected to automated sample preparation in a KingFisher™ Flex magnetic handling station using the novel approach based on MagReSyn<sup>®</sup> HILIC microspheres. For benchmarking, the pooled sample was also prepared using a published protocol based on an on-membrane (OM) protein capture and digestion workflow. Peptides were analyzed by LCMS in data-independent acquisition (DIA) mode using a Dionex Ultimate 3000 UPLC coupled to a Sciex 5600 mass spectrometer. The data were searched in Spectronaut™ 17. Both workflows showed similar peptide and protein identifications in the pooled sample. The uHLC workflow was easier to set up and complete, having less hands-on time than the OM method, with fewer manual processing steps. Lower peptide and protein coefficient of variation was observed in the uHLC technical replicates. Following statistical analysis, candidate protein markers were filtered, at ≥8.35-fold change in abundance, ≥2 unique peptides and ≤1% false discovery rate, and revealed 121 significant, differentially abundant proteins, some of which have known associations with kidney injury. The pilot data derived using this novel workflow provide information on the urinary proteome of patients with AKI. Further exploration in a larger cohort using this novel high-throughput method is warranted.</p>","PeriodicalId":20877,"journal":{"name":"Proteomes","volume":"11 4","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2023-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10594433/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/proteomes11040029","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
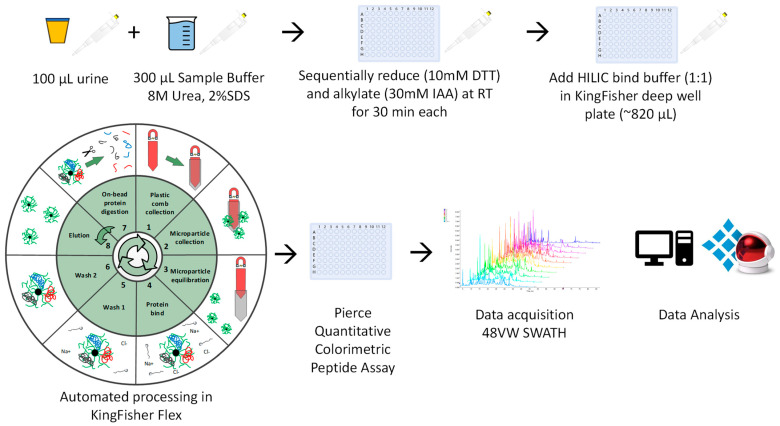
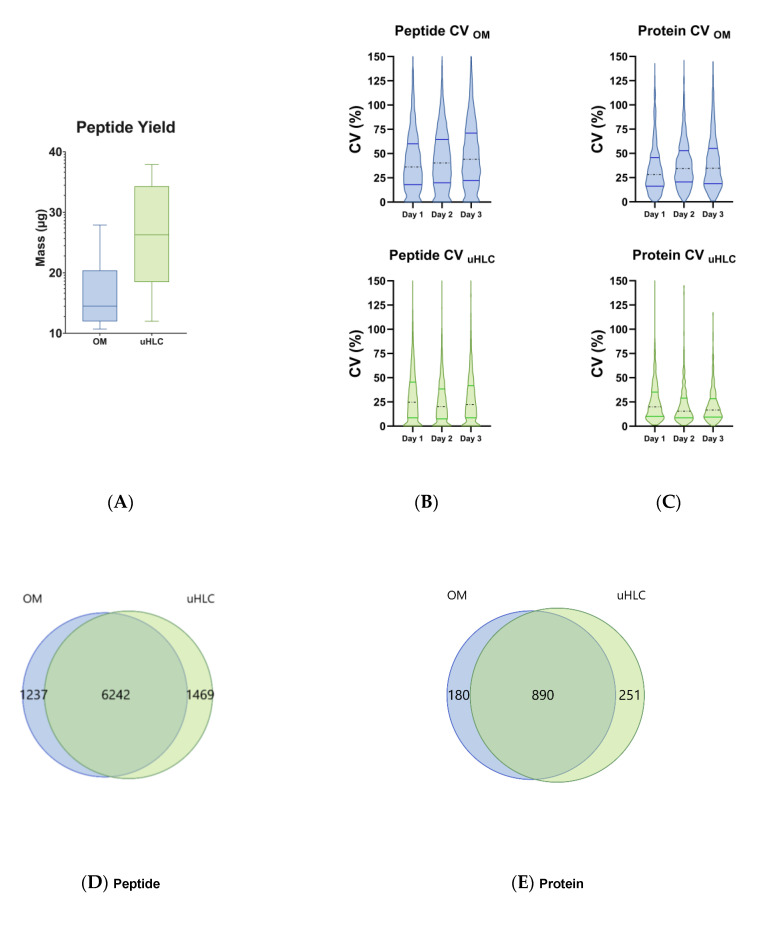
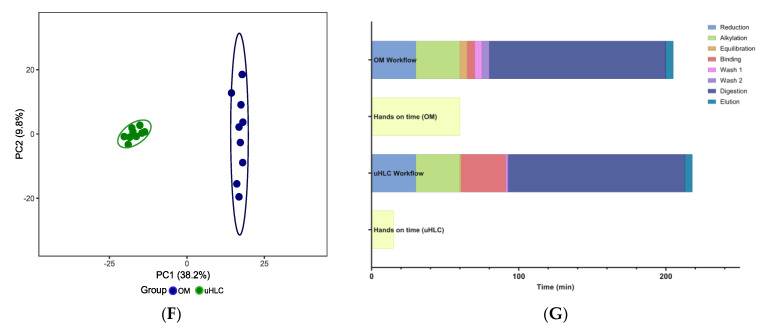
Urine provides a diverse source of information related to a patient's health status and is ideal for clinical proteomics due to its ease of collection. To date, most methods for the preparation of urine samples lack the throughput required to analyze large clinical cohorts. To this end, we developed a novel workflow, urine-HILIC (uHLC), based on an on-bead protein capture, clean-up, and digestion without the need for bottleneck processing steps such as protein precipitation or centrifugation. The workflow was applied to an acute kidney injury (AKI) pilot study. Urine from clinical samples and a pooled sample was subjected to automated sample preparation in a KingFisher™ Flex magnetic handling station using the novel approach based on MagReSyn® HILIC microspheres. For benchmarking, the pooled sample was also prepared using a published protocol based on an on-membrane (OM) protein capture and digestion workflow. Peptides were analyzed by LCMS in data-independent acquisition (DIA) mode using a Dionex Ultimate 3000 UPLC coupled to a Sciex 5600 mass spectrometer. The data were searched in Spectronaut™ 17. Both workflows showed similar peptide and protein identifications in the pooled sample. The uHLC workflow was easier to set up and complete, having less hands-on time than the OM method, with fewer manual processing steps. Lower peptide and protein coefficient of variation was observed in the uHLC technical replicates. Following statistical analysis, candidate protein markers were filtered, at ≥8.35-fold change in abundance, ≥2 unique peptides and ≤1% false discovery rate, and revealed 121 significant, differentially abundant proteins, some of which have known associations with kidney injury. The pilot data derived using this novel workflow provide information on the urinary proteome of patients with AKI. Further exploration in a larger cohort using this novel high-throughput method is warranted.
ProteomesBiochemistry, Genetics and Molecular Biology-Clinical Biochemistry
CiteScore
6.50
自引率
3.00%
发文量
37
审稿时长
11 weeks
期刊介绍:
Proteomes (ISSN 2227-7382) is an open access, peer reviewed journal on all aspects of proteome science. Proteomes covers the multi-disciplinary topics of structural and functional biology, protein chemistry, cell biology, methodology used for protein analysis, including mass spectrometry, protein arrays, bioinformatics, HTS assays, etc. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. Therefore, there is no restriction on the length of papers. Scope: -whole proteome analysis of any organism -disease/pharmaceutical studies -comparative proteomics -protein-ligand/protein interactions -structure/functional proteomics -gene expression -methodology -bioinformatics -applications of proteomics



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
