{"title":"The ground electronic state of ClF: Updated molecular constants and potential curves for 35ClF and 37ClF","authors":"Photos G. Hajigeorgiou","doi":"10.1016/j.jms.2023.111845","DOIUrl":null,"url":null,"abstract":"<div><p>A comprehensive assessment of available literature spectroscopic data and molecular constants for the <em>X</em> <sup>1</sup>Σ<sup>+</sup> ground electronic states of isotopologues <sup>35</sup>ClF and <sup>37</sup>ClF is undertaken. Three different approaches are employed in the analysis of the available information. The first approach involves merging molecular constants from various studies to yield an optimized set of Dunham coefficients {<em>Y</em><sub>01</sub>, <em>Y</em><sub>02</sub>, <em>Y</em><sub>03</sub>, <em>Y</em><sub>10</sub>, <em>Y</em><sub>11</sub>, <em>Y</em><sub>21</sub>, <em>Y</em><sub>02</sub> and <em>Y</em><sub>12</sub>}. Utilizing these updated constants, vibrational energies (<em>G<sub>υ</sub></em>) and rotational constants (<em>B<sub>υ</sub></em>) for <em>υ</em> = 0–9 are calculated, and RKR potentials are determined for both isotopologues. The second approach involves calculating synthetic spectroscopic line positions using literature molecular constants, with normally distributed random errors added on, and subjecting these to a modern direct-potential-fit analysis. This analysis produces a precise analytical potential energy function for <sup>35</sup>ClF, and Born-Oppenheimer breakdown functions that characterize adiabatic and non-adiabatic corrections. The third approach involves fitting the synthetic spectroscopic data directly to Dunham coefficients. The results of the three approaches are compared and discussed.</p></div>","PeriodicalId":16367,"journal":{"name":"Journal of Molecular Spectroscopy","volume":"398 ","pages":"Article 111845"},"PeriodicalIF":1.3000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Spectroscopy","FirstCategoryId":"101","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022285223001108","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, ATOMIC, MOLECULAR & CHEMICAL","Score":null,"Total":0}
引用次数: 0
Abstract
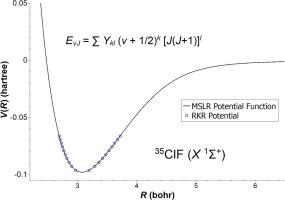
A comprehensive assessment of available literature spectroscopic data and molecular constants for the X1Σ+ ground electronic states of isotopologues 35ClF and 37ClF is undertaken. Three different approaches are employed in the analysis of the available information. The first approach involves merging molecular constants from various studies to yield an optimized set of Dunham coefficients {Y01, Y02, Y03, Y10, Y11, Y21, Y02 and Y12}. Utilizing these updated constants, vibrational energies (Gυ) and rotational constants (Bυ) for υ = 0–9 are calculated, and RKR potentials are determined for both isotopologues. The second approach involves calculating synthetic spectroscopic line positions using literature molecular constants, with normally distributed random errors added on, and subjecting these to a modern direct-potential-fit analysis. This analysis produces a precise analytical potential energy function for 35ClF, and Born-Oppenheimer breakdown functions that characterize adiabatic and non-adiabatic corrections. The third approach involves fitting the synthetic spectroscopic data directly to Dunham coefficients. The results of the three approaches are compared and discussed.
期刊介绍:
The Journal of Molecular Spectroscopy presents experimental and theoretical articles on all subjects relevant to molecular spectroscopy and its modern applications. An international medium for the publication of some of the most significant research in the field, the Journal of Molecular Spectroscopy is an invaluable resource for astrophysicists, chemists, physicists, engineers, and others involved in molecular spectroscopy research and practice.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
