{"title":"Mapping extended reaction coordinates in photochemical dynamics","authors":"Dave Townsend","doi":"10.1016/j.jms.2023.111807","DOIUrl":null,"url":null,"abstract":"<div><p>Laser-based experiments facilitate numerous strategies for interrogating the complex non-adiabatic dynamics operating in the excited states of molecules. Measurements may be broadly separated into frequency- and time-resolved variants, with a combination of different approaches (with different associated observables) typically being required to reveal a complete mechanistic picture. In the former category, quantum state-resolved information may often be obtained using narrow linewidth lasers. This provides detailed information relating to the starting point on the photochemical reaction coordinate (via the absorption spectrum) and the asymptotic endpoints (i.e. the photoproducts). Direct observation of the intermediate pathways connecting these two limits is often not possible, however, due to the inherently long temporal duration of the laser pulses relative to the typical timescales of non-adiabatic energy redistribution processes. It is therefore desirable to obtain complementary information that monitors real-time evolution along the reaction coordinate as excited state population traverses the potential energy landscape. This may be achieved in time-resolved pump–probe experiments conducted using ultrafast (i.e. sub-picosecond) laser pulses. The use of valence state photoionization for the probe step is a commonly employed methodology and has proved highly instructive in revealing subtle mechanistic details of key energy redistribution pathways operating in many different molecular systems. One frequent limitation here, however, is the restricted “view” along the reaction coordinate(s) connecting the initially prepared excited states to various photoproducts. Guided by examples drawn from recent work using time-resolved photoelectron imaging, this review will discuss such issues in detail and highlight some strategies that potentially help overcome them – with particular emphasis placed on the advantages of projecting as deeply as possible into the ionization continuum. The role of complementary measurements using other spectroscopic techniques and the importance of high-level supporting theory to guide data interpretation will also be reinforced.</p></div>","PeriodicalId":16367,"journal":{"name":"Journal of Molecular Spectroscopy","volume":"395 ","pages":"Article 111807"},"PeriodicalIF":1.3000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Spectroscopy","FirstCategoryId":"101","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022285223000723","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, ATOMIC, MOLECULAR & CHEMICAL","Score":null,"Total":0}
引用次数: 0
Abstract
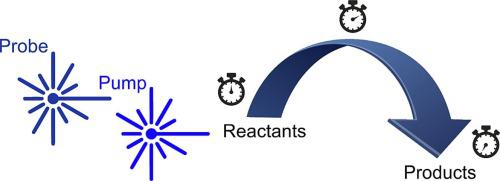
Laser-based experiments facilitate numerous strategies for interrogating the complex non-adiabatic dynamics operating in the excited states of molecules. Measurements may be broadly separated into frequency- and time-resolved variants, with a combination of different approaches (with different associated observables) typically being required to reveal a complete mechanistic picture. In the former category, quantum state-resolved information may often be obtained using narrow linewidth lasers. This provides detailed information relating to the starting point on the photochemical reaction coordinate (via the absorption spectrum) and the asymptotic endpoints (i.e. the photoproducts). Direct observation of the intermediate pathways connecting these two limits is often not possible, however, due to the inherently long temporal duration of the laser pulses relative to the typical timescales of non-adiabatic energy redistribution processes. It is therefore desirable to obtain complementary information that monitors real-time evolution along the reaction coordinate as excited state population traverses the potential energy landscape. This may be achieved in time-resolved pump–probe experiments conducted using ultrafast (i.e. sub-picosecond) laser pulses. The use of valence state photoionization for the probe step is a commonly employed methodology and has proved highly instructive in revealing subtle mechanistic details of key energy redistribution pathways operating in many different molecular systems. One frequent limitation here, however, is the restricted “view” along the reaction coordinate(s) connecting the initially prepared excited states to various photoproducts. Guided by examples drawn from recent work using time-resolved photoelectron imaging, this review will discuss such issues in detail and highlight some strategies that potentially help overcome them – with particular emphasis placed on the advantages of projecting as deeply as possible into the ionization continuum. The role of complementary measurements using other spectroscopic techniques and the importance of high-level supporting theory to guide data interpretation will also be reinforced.
期刊介绍:
The Journal of Molecular Spectroscopy presents experimental and theoretical articles on all subjects relevant to molecular spectroscopy and its modern applications. An international medium for the publication of some of the most significant research in the field, the Journal of Molecular Spectroscopy is an invaluable resource for astrophysicists, chemists, physicists, engineers, and others involved in molecular spectroscopy research and practice.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
