Asit Kumar Pradhan, Manaswini Ray, Venkatakrishnan Parthasarathy and Ashok Kumar Mishra
{"title":"Effects of donor and acceptor substituents on the photophysics of 4-ethynyl-2,1,3-benzothiadiazole derivatives†","authors":"Asit Kumar Pradhan, Manaswini Ray, Venkatakrishnan Parthasarathy and Ashok Kumar Mishra","doi":"10.1039/D3CP03318J","DOIUrl":null,"url":null,"abstract":"<p >The present work explores the photophysical, electrochemical, and fluorescence polarization properties of a group of π-conjugated phenylethynyl-2,1,3-benzothiadiazole derivatives (<strong>BTD</strong>s) bearing different electron-donating (ED) or electron-withdrawing (EW) substituents at the <em>para</em> position of the phenylethynyl moiety. The <strong>BTD</strong>s were synthesized through the Sonogashira cross-coupling reaction between 4-bromo-2,1,3-benzothiadiazole and the respective <em>para</em>-substituted phenylethynyl derivatives. The <strong>BTD</strong>s with the EW-substituents show relatively weak solvatochromic behavior, while the <strong>BTD</strong>s with the strong ED-substituents like methoxy and <em>N</em>,<em>N</em>-dimethylamino-based substituents (<strong>BTDPhOMe</strong> and <strong>BTDPhNMe<small><sub>2</sub></small></strong>) exhibit a pronounced solvatochromic behavior. The change in dipole moments in the excited states of the derivatives was calculated using Lippert–Mataga plots. The conclusions drawn on the spectral behavior of the molecules could be rationalized by TD-DFT calculations involving electron density difference (EDD) maps that correlate with the ICT characteristics of the molecules. The experimental and theoretical calculations reveal that the <strong>BTD</strong>s with the strong ED-substituents (strong push–pull type <strong>BTD</strong>s) have a strong ICT character in the excited state. These strong push–pull type <strong>BTD</strong>s show high fluorescence quantum yield (<em>Φ</em><small><sub>F</sub></small>) in apolar solvents and low <em>Φ</em><small><sub>F</sub></small> in polar solvents. In contrast, the <strong>BTD</strong>s with the weak ED-substituents (weak push–pull type <strong>BTD</strong>s) and EW-substituents (pull–pull type <strong>BTD</strong>s) have a weaker ICT character with low <em>Φ</em><small><sub>F</sub></small> in apolar and high <em>Φ</em><small><sub>F</sub></small> in polar solvent media. There is good a agreement among the HOMO–LUMO band gaps obtained from absorption spectroscopy and electrochemical studies and theoretical calculations. The fluorescence anisotropy measurement in the glycerol medium shows that the studied <strong>BTD</strong>s generally exhibit higher sensitivity towards microviscosity than the traditional <strong>DPH</strong> fluorescence anisotropy probe.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 42","pages":" 29327-29340"},"PeriodicalIF":2.9000,"publicationDate":"2023-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03318j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
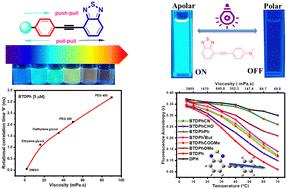
The present work explores the photophysical, electrochemical, and fluorescence polarization properties of a group of π-conjugated phenylethynyl-2,1,3-benzothiadiazole derivatives (BTDs) bearing different electron-donating (ED) or electron-withdrawing (EW) substituents at the para position of the phenylethynyl moiety. The BTDs were synthesized through the Sonogashira cross-coupling reaction between 4-bromo-2,1,3-benzothiadiazole and the respective para-substituted phenylethynyl derivatives. The BTDs with the EW-substituents show relatively weak solvatochromic behavior, while the BTDs with the strong ED-substituents like methoxy and N,N-dimethylamino-based substituents (BTDPhOMe and BTDPhNMe2) exhibit a pronounced solvatochromic behavior. The change in dipole moments in the excited states of the derivatives was calculated using Lippert–Mataga plots. The conclusions drawn on the spectral behavior of the molecules could be rationalized by TD-DFT calculations involving electron density difference (EDD) maps that correlate with the ICT characteristics of the molecules. The experimental and theoretical calculations reveal that the BTDs with the strong ED-substituents (strong push–pull type BTDs) have a strong ICT character in the excited state. These strong push–pull type BTDs show high fluorescence quantum yield (ΦF) in apolar solvents and low ΦF in polar solvents. In contrast, the BTDs with the weak ED-substituents (weak push–pull type BTDs) and EW-substituents (pull–pull type BTDs) have a weaker ICT character with low ΦF in apolar and high ΦF in polar solvent media. There is good a agreement among the HOMO–LUMO band gaps obtained from absorption spectroscopy and electrochemical studies and theoretical calculations. The fluorescence anisotropy measurement in the glycerol medium shows that the studied BTDs generally exhibit higher sensitivity towards microviscosity than the traditional DPH fluorescence anisotropy probe.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
