Xingyu Xie, Mingyang Shi, Xuying Zhou, Xianqiong Li, Gang Jiang and Jiguang Du
{"title":"Adsorption and diffusion of actinyls on the basal gibbsite (001) surface: a theoretical perspective†","authors":"Xingyu Xie, Mingyang Shi, Xuying Zhou, Xianqiong Li, Gang Jiang and Jiguang Du","doi":"10.1039/D3CP04088G","DOIUrl":null,"url":null,"abstract":"<p >Actinides are an important component of nuclear fuel for nuclear power and affect human health, and a key process in the transport of radionuclides in the environment is adsorption on mineral surfaces. In this work, we have used density functional theory (DFT) to investigate the microscopic adsorption and diffusion mechanisms of actinyls, U(<small>V</small>), U(<small>VI</small>), Np(<small>V</small>), Np(<small>VI</small>) Pu(<small>V</small>), and Pu(<small>VI</small>), on the gibbsite (001) surface. Actinyls(<small>VI</small>) are attached to the gibbsite surface through two An–O<small><sub>s</sub></small> bonds, which results in a bidentate inner sphere mode, while actinyls(<small>V</small>) favor a monodentate inner sphere adsorption mode with the gibbsite (001) surface. The solvent effects were considered through an explicit water cluster model. All the actinyls studied can be efficiently adsorbed on the gibbsite (001) surface with binding energies ranging from −113.9 kJ mol<small><sup>−1</sup></small> to −341.2 kJ mol<small><sup>−1</sup></small>. Electronic structure analyses indicate that the cooperation of the An–O<small><sub>s</sub></small> bonds and hydrogen bonds leads to high adsorption stability of the actinyls with the gibbsite surface. The diffusion barriers of the actinyls on the gibbsite surface were determined, and the high energy barriers indicate that this type of gas-phase diffusion process is not likely to take place.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 43","pages":" 29680-29689"},"PeriodicalIF":2.9000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp04088g","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
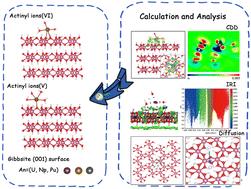
Actinides are an important component of nuclear fuel for nuclear power and affect human health, and a key process in the transport of radionuclides in the environment is adsorption on mineral surfaces. In this work, we have used density functional theory (DFT) to investigate the microscopic adsorption and diffusion mechanisms of actinyls, U(V), U(VI), Np(V), Np(VI) Pu(V), and Pu(VI), on the gibbsite (001) surface. Actinyls(VI) are attached to the gibbsite surface through two An–Os bonds, which results in a bidentate inner sphere mode, while actinyls(V) favor a monodentate inner sphere adsorption mode with the gibbsite (001) surface. The solvent effects were considered through an explicit water cluster model. All the actinyls studied can be efficiently adsorbed on the gibbsite (001) surface with binding energies ranging from −113.9 kJ mol−1 to −341.2 kJ mol−1. Electronic structure analyses indicate that the cooperation of the An–Os bonds and hydrogen bonds leads to high adsorption stability of the actinyls with the gibbsite surface. The diffusion barriers of the actinyls on the gibbsite surface were determined, and the high energy barriers indicate that this type of gas-phase diffusion process is not likely to take place.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
