Corina Ramona Nicolescu, Clara Cremillieux, Jean-Louis Stephan
{"title":"Duodenogastric Intussusception in a 14-Week-Old Infant with Donohue Syndrome: Case Study.","authors":"Corina Ramona Nicolescu, Clara Cremillieux, Jean-Louis Stephan","doi":"10.1155/2023/7799234","DOIUrl":null,"url":null,"abstract":"<p><p>Donohue syndrome (DS) is a rare recessively inherited disorder characterized by severe insulin resistance caused by genetic defects affecting the insulin receptor. The classical clinical characteristics include severe intrauterine growth restriction, craniofacial dysmorphic features, body and skin features, and soft tissue overgrowth. Postnatal growth retardation, cardiac, gastrointestinal, and renal complications, and infection susceptibility develop within the first few months of life, leading to a short life expectancy (<2 years). The classical metabolic abnormalities vary from fasting hypoglycemia to postprandial hyperglycemia with severe hyperinsulinemia. We present the case of a 14-week-old infant with DS who developed cardiac, renal, hepatic, pancreatic, and gastrointestinal features, all of them previously reported in infants with DS. The gastrointestinal features started during the first week of life and included abdominal distension, feeding difficulties, intermittent vomiting, and two episodes of intestinal obstruction. The diagnosis of duodenogastric intussusception was made, and this previously unreported complication tragically resulted in mortality. We discuss how basic mechanisms of cross-talk between insulin and insulin-growth factor 1 receptors could be linked to hyperinsulinemia and its associated comorbidities.</p>","PeriodicalId":9623,"journal":{"name":"Case Reports in Pediatrics","volume":"2023 ","pages":"7799234"},"PeriodicalIF":0.5000,"publicationDate":"2023-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10599843/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/7799234","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
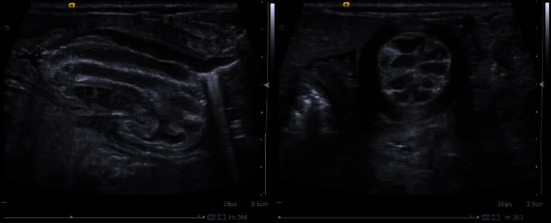
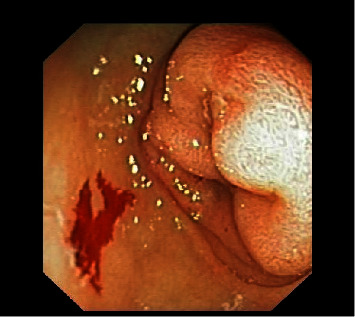
Donohue syndrome (DS) is a rare recessively inherited disorder characterized by severe insulin resistance caused by genetic defects affecting the insulin receptor. The classical clinical characteristics include severe intrauterine growth restriction, craniofacial dysmorphic features, body and skin features, and soft tissue overgrowth. Postnatal growth retardation, cardiac, gastrointestinal, and renal complications, and infection susceptibility develop within the first few months of life, leading to a short life expectancy (<2 years). The classical metabolic abnormalities vary from fasting hypoglycemia to postprandial hyperglycemia with severe hyperinsulinemia. We present the case of a 14-week-old infant with DS who developed cardiac, renal, hepatic, pancreatic, and gastrointestinal features, all of them previously reported in infants with DS. The gastrointestinal features started during the first week of life and included abdominal distension, feeding difficulties, intermittent vomiting, and two episodes of intestinal obstruction. The diagnosis of duodenogastric intussusception was made, and this previously unreported complication tragically resulted in mortality. We discuss how basic mechanisms of cross-talk between insulin and insulin-growth factor 1 receptors could be linked to hyperinsulinemia and its associated comorbidities.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
