Belinda Hernández , Stephen R Pennington , Andrew C Parnell
{"title":"Bayesian methods for proteomic biomarker development","authors":"Belinda Hernández , Stephen R Pennington , Andrew C Parnell","doi":"10.1016/j.euprot.2015.08.001","DOIUrl":null,"url":null,"abstract":"<div><p>The advent of liquid chromatography mass spectrometry has seen a dramatic increase in the amount of data derived from proteomic biomarker discovery. These experiments have seemingly identified many potential candidate biomarkers. Frustratingly, very few of these candidates have been evaluated and validated sufficiently such that that they have progressed to the stage of routine clinical use. It is becoming apparent that the statistical methods used to evaluate the performance of new candidate biomarkers are a major limitation in their development. Bayesian methods offer some advantages over traditional statistical and machine learning methods. In particular they can incorporate external information into current experiments so as to guide biomarker selection. Further, they can be more robust to over-fitting than other approaches, especially when the number of samples used for discovery is relatively small.</p><p>In this review we provide an introduction to Bayesian inference and demonstrate some of the advantages of using a Bayesian framework. We summarize how Bayesian methods have been used previously in proteomics and other areas of bioinformatics. Finally, we describe some popular and emerging Bayesian models from the statistical literature and provide a worked tutorial including code snippets to show how these methods may be applied for the evaluation of proteomic biomarkers.</p></div>","PeriodicalId":38260,"journal":{"name":"EuPA Open Proteomics","volume":"9 ","pages":"Pages 54-64"},"PeriodicalIF":0.0000,"publicationDate":"2015-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.euprot.2015.08.001","citationCount":"18","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EuPA Open Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2212968515300180","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/8/10 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 18
Abstract
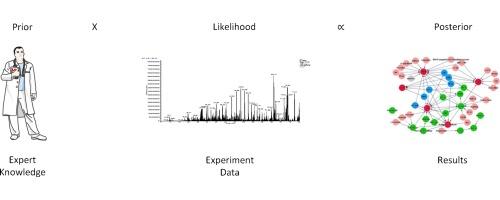
The advent of liquid chromatography mass spectrometry has seen a dramatic increase in the amount of data derived from proteomic biomarker discovery. These experiments have seemingly identified many potential candidate biomarkers. Frustratingly, very few of these candidates have been evaluated and validated sufficiently such that that they have progressed to the stage of routine clinical use. It is becoming apparent that the statistical methods used to evaluate the performance of new candidate biomarkers are a major limitation in their development. Bayesian methods offer some advantages over traditional statistical and machine learning methods. In particular they can incorporate external information into current experiments so as to guide biomarker selection. Further, they can be more robust to over-fitting than other approaches, especially when the number of samples used for discovery is relatively small.
In this review we provide an introduction to Bayesian inference and demonstrate some of the advantages of using a Bayesian framework. We summarize how Bayesian methods have been used previously in proteomics and other areas of bioinformatics. Finally, we describe some popular and emerging Bayesian models from the statistical literature and provide a worked tutorial including code snippets to show how these methods may be applied for the evaluation of proteomic biomarkers.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
