{"title":"Quantitative analysis of high-throughput biological data","authors":"Hsueh-Fen Juan, Hsuan-Cheng Huang","doi":"10.1002/wcms.1658","DOIUrl":null,"url":null,"abstract":"<p>The study of multiple “omes,” such as the genome, transcriptome, proteome, and metabolome has become widespread in biomedical research. High-throughput techniques enable the rapid generation of high-dimensional multiomics data. This multiomics approach provides a more complete perspective to study biological systems compared with traditional methods. However, the quantitative analysis and integration of distinct types of high-dimensional omics data remain a challenge. Here, we provide an up-to-date and comprehensive review of the methods used for omics data quantification and integration. We first review the quantitative analysis of not only bulk but also single-cell transcriptomics data, as well as proteomics data. Current methods for reducing batch effects and integrating heterogeneous high-dimensional data are then introduced. Network analysis on large-scale biomedical data can capture the global properties of drugs, targets, and disease relationships, thus enabling a better understanding of biological systems. Current trends in the applications and methods used to extend quantitative omics data analysis to biological networks are also discussed.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 4","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1658","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 1
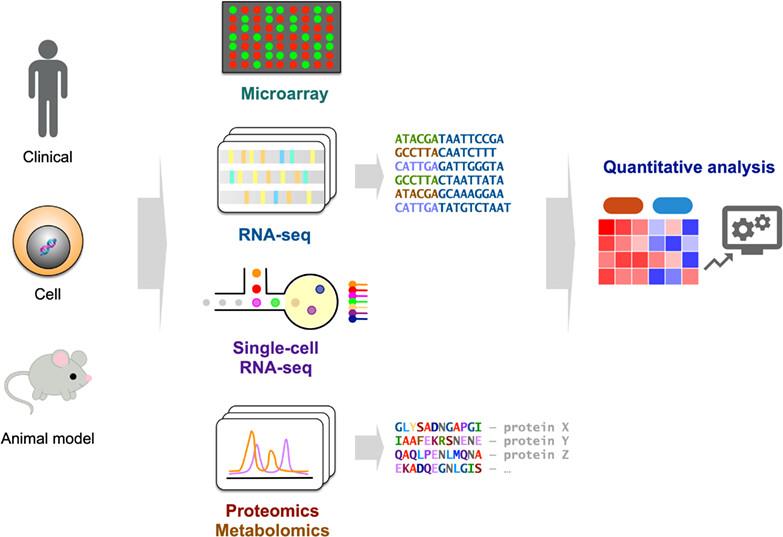
Abstract
The study of multiple “omes,” such as the genome, transcriptome, proteome, and metabolome has become widespread in biomedical research. High-throughput techniques enable the rapid generation of high-dimensional multiomics data. This multiomics approach provides a more complete perspective to study biological systems compared with traditional methods. However, the quantitative analysis and integration of distinct types of high-dimensional omics data remain a challenge. Here, we provide an up-to-date and comprehensive review of the methods used for omics data quantification and integration. We first review the quantitative analysis of not only bulk but also single-cell transcriptomics data, as well as proteomics data. Current methods for reducing batch effects and integrating heterogeneous high-dimensional data are then introduced. Network analysis on large-scale biomedical data can capture the global properties of drugs, targets, and disease relationships, thus enabling a better understanding of biological systems. Current trends in the applications and methods used to extend quantitative omics data analysis to biological networks are also discussed.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
