Mitochondria-targeted atovaquone promotes anti-lung cancer immunity by reshaping tumor microenvironment and enhancing energy metabolism of anti-tumor immune cells
Donghai Xiong, Zheng Yin, Mofei Huang, Yian Wang, Micael Hardy, Balaraman Kalyanaraman, Stephen T Wong, Ming You
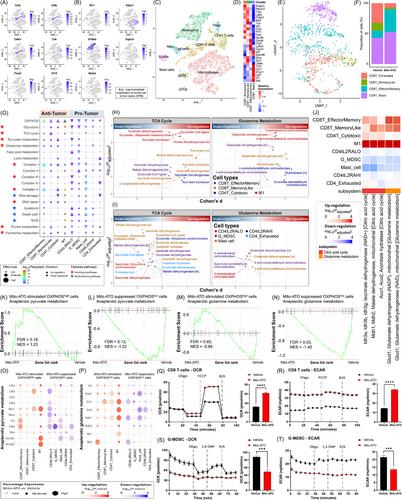
{"title":"Mitochondria-targeted atovaquone promotes anti-lung cancer immunity by reshaping tumor microenvironment and enhancing energy metabolism of anti-tumor immune cells","authors":"Donghai Xiong, Zheng Yin, Mofei Huang, Yian Wang, Micael Hardy, Balaraman Kalyanaraman, Stephen T Wong, Ming You","doi":"10.1002/cac2.12500","DOIUrl":null,"url":null,"abstract":"<p>Atovaquone (ATO), a mitochondrial inhibitor, has anti-cancer effects [<span>1</span>]. Based on ATO, we developed mitochondria-targeted atovaquone (Mito-ATO) that had even stronger anti-tumor efficacy than ATO [<span>2</span>]. We synthesized Mito-ATO by attaching the bulky triphenylphosphonium (TPP) group to ATO via a ten-carbon alkyl chain (Supplementary file of methods; Supplementary Figure S1). To assess the effects of Mito-ATO on tumor microenvironment, we conducted single-cell RNA-sequencing (scRNA-seq) on treated immune cells from mice having lung tumors either treated with or without Mito-ATO. Seurat was used for clustering and annotation of CD45<sup>+</sup> immune cells [<span>3</span>]. The detected lymphoid cell populations were CD8<sup>+</sup> T cells, CD4<sup>+</sup> T cells, regulatory T cells (Tregs), gamma-delta T (Tgd) cells, B cells, and natural killer (NK) cells; and the myeloid cells identified were macrophages, neutrophils, plasmacytoid dendritic cells (pDCs), conventional dendritic cells (cDCs) and mast cells (Figure 1A-C). Clustering of CD4<sup>+</sup> T cells into seven subpopulations, the separation of neutrophils and granulocytic myeloid-derived suppressor cells (G-MDSCs), and the division of macrophages into M1 and M2 subtypes were described in our previous publication [<span>2</span>]. In this study, we further divided CD8<sup>+</sup> T cells into four subpopulations, i.e., exhausted CD8<sup>+</sup> T (CD8T_Exhausted) cells, memory like CD8<sup>+</sup> T (CD8T_MemoryLike) cells, effector memory like CD8<sup>+</sup> T (CD8T_EffectorMemory) cells and naive CD8<sup>+</sup> T (CD8T_Naive) cells, using the tumor-infiltrating CD8<sup>+</sup> lymphocyte state predictor (TILPRED) method [<span>4</span>] (Figure 1D-E). Probability scores computed with TILPRED could discriminate CD8T_Exhausted from CD8T_MemoryLike cells despite overlap between the two subsets on UMAP representation (Supplementary Figure S2). Mito-ATO treatment significantly decreased the proportion of the CD8T_Exhausted cells (7.3% vs. 32.5%, <i>P</i> < 0.001) but increased the proportion of anti-tumor CD8T_EffectorMemory cells as compared with vehicle treatment (37.3% vs. 11.9%, <i>P</i> < 0.001) (Figure 1F). In comparison, the percentages of CD8<sup>+</sup> T cells out of total T cells were not different between the two groups (Supplementary Table S1). For validation, we verified that Mito-ATO treatment induced changes in CD8<sup>+</sup> T cell repartition by conducting flow cytometry. Mito-ATO treatment significantly increased the percentage of cytotoxic tumor necrosis factor-alpha (TNF-α)<sup>+</sup>CD8<sup>+</sup> T cells and decreased the percentage of programmed cell death protein-1 (PD-1)<sup>+</sup> T cell immunoglobulin and mucin domain-containing protein 3 (TIM3)<sup>+</sup>CD8<sup>+</sup> T cells (Supplementary Figure S3). These matched the scRNA-seq results. We also observed a slight trend toward the upregulation of genes involved in CD8<sup>+</sup> T cell recruitment: Ccl25, Ccr7, Cxcl10, Cxcr3, Icam1, and S1pr1 (Supplementary Figure S4).</p><p>Mito-ATO treatment significantly up-regulated oxidative phosphorylation (OXPHOS) activity in four anti-tumor immune cell populations, i.e., CD8T_EffectorMemory cells, CD8T_MemoryLike cells, cytotoxic CD4<sup>+</sup> T cells (CD4T_Cytotoxic), and M1 macrophage cells (Figure 1G). Particularly, the upregulated genes after Mito-ATO treatment were significantly enriched for T cell differentiation (Supplementary Figure S5), suggesting that Mito-ATO treatment may induce CD8<sup>+</sup> T cell differentiation. In contrast, Mito-ATO treatment significantly down-regulated OXPHOS activity in five pro-tumor immune cell populations, i.e., interleukin-2 receptor subunit alpha (IL2RA)-low CD4<sup>+</sup> (CD4IL2RALO) Tregs, G-MDSCs, mast cells, IL2RA-high CD4<sup>+</sup> (CD4IL2RAHI) Tregs, and exhausted CD4<sup>+</sup> T cells (CD4T_Exhausted) (Figure 1G). The two types of Treg cells were named following the previous practice [<span>2, 5</span>]. Ten metabolic pathways activity changes were similar to OXPHOS activity changes by Mito-ATO treatment in the above immune cell populations. These pathways were glycolysis, the tricarboxylic acid (TCA) cycle, pyruvate metabolism, glutamine metabolism, Complex I, Complex III, Complex V, DNA repair, purine metabolism and pyrimidine metabolism (Figure 1G). The changes of four cell death-related pathways, i.e., DNA damage, apoptosis, cell death, and reactive oxygen species (ROS) pathways, were negatively correlated with OXPHOS activity changes by Mito-ATO treatment (Figure 1G). These suggested that Mito-ATO enhances energy metabolism and suppresses cell death in anti-tumor immune cells while inhibiting energy metabolism and promoting cell death in pro-tumor immune cells.</p><p>Compass metabolism analysis [<span>6</span>] showed similar results (Supplementary Figures S6-S14) . The key metabolic reactions of the TCA cycle and glutamine metabolism were up-regulated in the anti-tumor immune cell populations but down-regulated in the pro-tumor immune cell populations (Figure 1H-I). We identified key metabolic reactions differentially regulated by Mito-ATO across anti-tumor and pro-tumor immune cells, including aconitase 1 (Aco1)/Aco2, malate dehydrogenase 1 (Mdh1)/Mdh2, isocitrate dehydrogenase 3 alpha (Idh3a)/Idh3b/Idh3g in TCA cycle and the glutamate dehydrogenase 1 (Glud1) enzyme in glutamine metabolism. These metabolic reactions were consistently up-regulated by Mito-ATO in anti-tumor immune cells but down-regulated in pro-tumor immune cells (Figure 1J).</p><p>Furthermore, we performed gene set enrichment analysis (GSEA) [<span>7</span>]. We combined datasets of anti-tumor immune cells, which were termed Mito-ATO-stimulated OXPHOS-high cells since their OXPHOS was enhanced by Mito-ATO treatment. Analogously, we combined the pro-tumor immune cells and named them Mito-ATO-suppressed OXPHOS-low cells. The pyruvate pathway was up-regulated by Mito-ATO in Mito-ATO-stimulated OXPHOS-high cells (false discovery rate [FDR] = 0.18, normalized enrichment score [NES] = 1.23, Figure 1K) while down-regulated in Mito-ATO-suppressed OXPHOS-low cells (FDR = 0.13, NES = -1.22) (Figure 1L). The glutamine pathway was not significantly changed by Mito-ATO in Mito-ATO-stimulated OXPHOS-high cells (FDR = 0.65, NES = -0.86, Figure 1M) but down-regulated in Mito-ATO-suppressed OXPHOS-low cells (FDR = 0.03, NES = -1.40, Figure 1N). For pyruvate metabolism, Mito-ATO treatment up-regulated lactate dehydrogenase A (Ldha), mitochondrial pyruvate carrier 2 (Mpc2), pyruvate kinase (Pkm) expression in Mito-ATO-stimulated OXPHOS-high cells while down-regulated their expression in Mito-ATO-suppressed OXPHOS-low cells (Figure 1O). For the glutamine metabolism pathway, Mito-ATO treatment up-regulated Glud1, glutamic-oxaloacetic transaminase 1 (Got1) and Mdh1 expression in Mito-ATO-stimulated OXPHOS-high cells while down-regulated their expression in Mito-ATO-suppressed OXPHOS-low cells (Figure 1P). Interestingly, Mdh1 and Glud1 were also identified by Compass analysis to be significant TCA cycle and glutamine metabolic reaction regulators (Figure 1J). Therefore, the anti-cancer efficacy of Mito-ATO treatment may be realized through differential regulation of TCA and glutamine metabolism between anti-tumor and pro-tumor immune cells, in which the expression changes of Mdh1 and Glud1 could be critical. As orthogonal validation, the Seahorse metabolic flux assay showed that Mito-ATO treatment significantly increased OXPHOS activity and aerobic glycolysis in activated CD8<sup>+</sup> T (Figure 1Q-R). In contrast, Mito-ATO significantly suppressed OXPHOS and glycolysis in G-MDSCs (Figure 1S-T). These supported the predicted higher OXPHOS and glycolysis in effector memory CD8<sup>+</sup> T cells and lower OXPHOS and glycolysis in G-MDSCs upon Mito-ATO treatment.</p><p>The metabolic plasticity of different types of cells in the tumor microenvironment (TME) in response to a glutaminase inhibitor Ethyl 2-(2-amino-4-methylpentanamido)-DON (JHU083) has been reported [<span>8</span>]. In the present study, we found that Mito-ATO may differentially regulate pyruvate metabolism, glutamine metabolism and TCA cycle across immune cells with distinct roles in the TME. The metabolic plasticity effects caused by Mito-ATO treatment may contribute to the overall efficacy of this drug on lung tumors. Mito-ATO's parental compound – ATO has begun to be applied to anti-cancer clinical trials [<span>9</span>]. Given that Mito-ATO is much more potent against human cancer cell lines compared to ATO [<span>10</span>], it is reasonable to predict that Mito-ATO has great potential in clinics.</p><p>DHX did the data analysis of this project and drafted the manuscript. ZY helped with Compass analysis. MFH did the mice experiment and scRNA-seq experiment. MH synthesized the Mito-ATO compound used in this study. YW, BK, and STW helped in preparing experimental samples and worked with MY to revise the manuscript. MY also designed this study.</p><p>The authors declare no conflict of Interest.</p><p>This research was supported by National Institutes of Health (NIH): R01CA223804, R01CA232433, R01CA205633, and R01CA280746.</p><p>The animal study was reviewed, and all procedures were approved by the Medical College of Wisconsin (MCW) Institutional Animal Care and Use Committee (Ethics approval number of AUA00001807).</p><p>Not applicable.</p>","PeriodicalId":9495,"journal":{"name":"Cancer Communications","volume":"44 3","pages":"448-452"},"PeriodicalIF":24.9000,"publicationDate":"2023-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cac2.12500","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Communications","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/cac2.12500","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Atovaquone (ATO), a mitochondrial inhibitor, has anti-cancer effects [1]. Based on ATO, we developed mitochondria-targeted atovaquone (Mito-ATO) that had even stronger anti-tumor efficacy than ATO [2]. We synthesized Mito-ATO by attaching the bulky triphenylphosphonium (TPP) group to ATO via a ten-carbon alkyl chain (Supplementary file of methods; Supplementary Figure S1). To assess the effects of Mito-ATO on tumor microenvironment, we conducted single-cell RNA-sequencing (scRNA-seq) on treated immune cells from mice having lung tumors either treated with or without Mito-ATO. Seurat was used for clustering and annotation of CD45+ immune cells [3]. The detected lymphoid cell populations were CD8+ T cells, CD4+ T cells, regulatory T cells (Tregs), gamma-delta T (Tgd) cells, B cells, and natural killer (NK) cells; and the myeloid cells identified were macrophages, neutrophils, plasmacytoid dendritic cells (pDCs), conventional dendritic cells (cDCs) and mast cells (Figure 1A-C). Clustering of CD4+ T cells into seven subpopulations, the separation of neutrophils and granulocytic myeloid-derived suppressor cells (G-MDSCs), and the division of macrophages into M1 and M2 subtypes were described in our previous publication [2]. In this study, we further divided CD8+ T cells into four subpopulations, i.e., exhausted CD8+ T (CD8T_Exhausted) cells, memory like CD8+ T (CD8T_MemoryLike) cells, effector memory like CD8+ T (CD8T_EffectorMemory) cells and naive CD8+ T (CD8T_Naive) cells, using the tumor-infiltrating CD8+ lymphocyte state predictor (TILPRED) method [4] (Figure 1D-E). Probability scores computed with TILPRED could discriminate CD8T_Exhausted from CD8T_MemoryLike cells despite overlap between the two subsets on UMAP representation (Supplementary Figure S2). Mito-ATO treatment significantly decreased the proportion of the CD8T_Exhausted cells (7.3% vs. 32.5%, P < 0.001) but increased the proportion of anti-tumor CD8T_EffectorMemory cells as compared with vehicle treatment (37.3% vs. 11.9%, P < 0.001) (Figure 1F). In comparison, the percentages of CD8+ T cells out of total T cells were not different between the two groups (Supplementary Table S1). For validation, we verified that Mito-ATO treatment induced changes in CD8+ T cell repartition by conducting flow cytometry. Mito-ATO treatment significantly increased the percentage of cytotoxic tumor necrosis factor-alpha (TNF-α)+CD8+ T cells and decreased the percentage of programmed cell death protein-1 (PD-1)+ T cell immunoglobulin and mucin domain-containing protein 3 (TIM3)+CD8+ T cells (Supplementary Figure S3). These matched the scRNA-seq results. We also observed a slight trend toward the upregulation of genes involved in CD8+ T cell recruitment: Ccl25, Ccr7, Cxcl10, Cxcr3, Icam1, and S1pr1 (Supplementary Figure S4).
Mito-ATO treatment significantly up-regulated oxidative phosphorylation (OXPHOS) activity in four anti-tumor immune cell populations, i.e., CD8T_EffectorMemory cells, CD8T_MemoryLike cells, cytotoxic CD4+ T cells (CD4T_Cytotoxic), and M1 macrophage cells (Figure 1G). Particularly, the upregulated genes after Mito-ATO treatment were significantly enriched for T cell differentiation (Supplementary Figure S5), suggesting that Mito-ATO treatment may induce CD8+ T cell differentiation. In contrast, Mito-ATO treatment significantly down-regulated OXPHOS activity in five pro-tumor immune cell populations, i.e., interleukin-2 receptor subunit alpha (IL2RA)-low CD4+ (CD4IL2RALO) Tregs, G-MDSCs, mast cells, IL2RA-high CD4+ (CD4IL2RAHI) Tregs, and exhausted CD4+ T cells (CD4T_Exhausted) (Figure 1G). The two types of Treg cells were named following the previous practice [2, 5]. Ten metabolic pathways activity changes were similar to OXPHOS activity changes by Mito-ATO treatment in the above immune cell populations. These pathways were glycolysis, the tricarboxylic acid (TCA) cycle, pyruvate metabolism, glutamine metabolism, Complex I, Complex III, Complex V, DNA repair, purine metabolism and pyrimidine metabolism (Figure 1G). The changes of four cell death-related pathways, i.e., DNA damage, apoptosis, cell death, and reactive oxygen species (ROS) pathways, were negatively correlated with OXPHOS activity changes by Mito-ATO treatment (Figure 1G). These suggested that Mito-ATO enhances energy metabolism and suppresses cell death in anti-tumor immune cells while inhibiting energy metabolism and promoting cell death in pro-tumor immune cells.
Compass metabolism analysis [6] showed similar results (Supplementary Figures S6-S14) . The key metabolic reactions of the TCA cycle and glutamine metabolism were up-regulated in the anti-tumor immune cell populations but down-regulated in the pro-tumor immune cell populations (Figure 1H-I). We identified key metabolic reactions differentially regulated by Mito-ATO across anti-tumor and pro-tumor immune cells, including aconitase 1 (Aco1)/Aco2, malate dehydrogenase 1 (Mdh1)/Mdh2, isocitrate dehydrogenase 3 alpha (Idh3a)/Idh3b/Idh3g in TCA cycle and the glutamate dehydrogenase 1 (Glud1) enzyme in glutamine metabolism. These metabolic reactions were consistently up-regulated by Mito-ATO in anti-tumor immune cells but down-regulated in pro-tumor immune cells (Figure 1J).
Furthermore, we performed gene set enrichment analysis (GSEA) [7]. We combined datasets of anti-tumor immune cells, which were termed Mito-ATO-stimulated OXPHOS-high cells since their OXPHOS was enhanced by Mito-ATO treatment. Analogously, we combined the pro-tumor immune cells and named them Mito-ATO-suppressed OXPHOS-low cells. The pyruvate pathway was up-regulated by Mito-ATO in Mito-ATO-stimulated OXPHOS-high cells (false discovery rate [FDR] = 0.18, normalized enrichment score [NES] = 1.23, Figure 1K) while down-regulated in Mito-ATO-suppressed OXPHOS-low cells (FDR = 0.13, NES = -1.22) (Figure 1L). The glutamine pathway was not significantly changed by Mito-ATO in Mito-ATO-stimulated OXPHOS-high cells (FDR = 0.65, NES = -0.86, Figure 1M) but down-regulated in Mito-ATO-suppressed OXPHOS-low cells (FDR = 0.03, NES = -1.40, Figure 1N). For pyruvate metabolism, Mito-ATO treatment up-regulated lactate dehydrogenase A (Ldha), mitochondrial pyruvate carrier 2 (Mpc2), pyruvate kinase (Pkm) expression in Mito-ATO-stimulated OXPHOS-high cells while down-regulated their expression in Mito-ATO-suppressed OXPHOS-low cells (Figure 1O). For the glutamine metabolism pathway, Mito-ATO treatment up-regulated Glud1, glutamic-oxaloacetic transaminase 1 (Got1) and Mdh1 expression in Mito-ATO-stimulated OXPHOS-high cells while down-regulated their expression in Mito-ATO-suppressed OXPHOS-low cells (Figure 1P). Interestingly, Mdh1 and Glud1 were also identified by Compass analysis to be significant TCA cycle and glutamine metabolic reaction regulators (Figure 1J). Therefore, the anti-cancer efficacy of Mito-ATO treatment may be realized through differential regulation of TCA and glutamine metabolism between anti-tumor and pro-tumor immune cells, in which the expression changes of Mdh1 and Glud1 could be critical. As orthogonal validation, the Seahorse metabolic flux assay showed that Mito-ATO treatment significantly increased OXPHOS activity and aerobic glycolysis in activated CD8+ T (Figure 1Q-R). In contrast, Mito-ATO significantly suppressed OXPHOS and glycolysis in G-MDSCs (Figure 1S-T). These supported the predicted higher OXPHOS and glycolysis in effector memory CD8+ T cells and lower OXPHOS and glycolysis in G-MDSCs upon Mito-ATO treatment.
The metabolic plasticity of different types of cells in the tumor microenvironment (TME) in response to a glutaminase inhibitor Ethyl 2-(2-amino-4-methylpentanamido)-DON (JHU083) has been reported [8]. In the present study, we found that Mito-ATO may differentially regulate pyruvate metabolism, glutamine metabolism and TCA cycle across immune cells with distinct roles in the TME. The metabolic plasticity effects caused by Mito-ATO treatment may contribute to the overall efficacy of this drug on lung tumors. Mito-ATO's parental compound – ATO has begun to be applied to anti-cancer clinical trials [9]. Given that Mito-ATO is much more potent against human cancer cell lines compared to ATO [10], it is reasonable to predict that Mito-ATO has great potential in clinics.
DHX did the data analysis of this project and drafted the manuscript. ZY helped with Compass analysis. MFH did the mice experiment and scRNA-seq experiment. MH synthesized the Mito-ATO compound used in this study. YW, BK, and STW helped in preparing experimental samples and worked with MY to revise the manuscript. MY also designed this study.
The authors declare no conflict of Interest.
This research was supported by National Institutes of Health (NIH): R01CA223804, R01CA232433, R01CA205633, and R01CA280746.
The animal study was reviewed, and all procedures were approved by the Medical College of Wisconsin (MCW) Institutional Animal Care and Use Committee (Ethics approval number of AUA00001807).
期刊介绍:
Cancer Communications is an open access, peer-reviewed online journal that encompasses basic, clinical, and translational cancer research. The journal welcomes submissions concerning clinical trials, epidemiology, molecular and cellular biology, and genetics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
