Pimchanok Kulsirichawaroj, Yanin Suksangkharn, Da Eun Nam, Theeraphong Pho-Iam, Chanin Limwongse, Ki Wha Chung, Oranee Sanmaneechai, Stephan L Zuchner, Byung-Ok Choi
{"title":"Gene Distribution in Pediatric-Onset Inherited Peripheral Neuropathy: A Single Tertiary Center in Thailand.","authors":"Pimchanok Kulsirichawaroj, Yanin Suksangkharn, Da Eun Nam, Theeraphong Pho-Iam, Chanin Limwongse, Ki Wha Chung, Oranee Sanmaneechai, Stephan L Zuchner, Byung-Ok Choi","doi":"10.3233/JND-230174","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Inherited peripheral neuropathy presents a diagnostic and therapeutic challenge due to its association with mutations in over 100 genes. This condition leads to long-term disability and poses a substantial healthcare burden on society.</p><p><strong>Objective: </strong>This study aimed to investigate the distribution of genes and establish the genotype-phenotype correlations, focusing on pediatric-onset cases.</p><p><strong>Methods: </strong>Exome sequencing and other analytical techniques were employed to identify pathogenic variants, including duplication analysis of the PMP22 gene. Each patient underwent physical examination and electrophysiological studies. Genotypes were correlated with phenotypic features, such as age at disease onset and ulnar motor nerve conduction velocity.</p><p><strong>Results: </strong>We identified 35 patients with pediatric-onset inherited peripheral neuropathy. Pathogenic or likely pathogenic variants were confirmed in 24 out of 35 (68.6%) patients, with 4 of these variants being novel. A confirmed molecular diagnosis was achieved in 90.9% (10/11) of patients with demyelinating Charcot-Marie-Tooth disease (CMT) and 56.3% (9/16) of patients with axonal CMT. Among patients with infantile-onset CMT (≤2 years), the most common causative genes were MFN2 and NEFL, while GDAP1 and MFN2 were frequent causes among patients with childhood- or adolescent-onset CMT (3-9 years).</p><p><strong>Conclusions: </strong>The MFN2 gene was the most commonly implicated gene, and the axonal type was predominant in this cohort of Thai patients with pediatric-onset inherited peripheral neuropathy.</p>","PeriodicalId":16536,"journal":{"name":"Journal of neuromuscular diseases","volume":" ","pages":"191-199"},"PeriodicalIF":3.4000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10789325/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of neuromuscular diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3233/JND-230174","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Inherited peripheral neuropathy presents a diagnostic and therapeutic challenge due to its association with mutations in over 100 genes. This condition leads to long-term disability and poses a substantial healthcare burden on society.
Objective: This study aimed to investigate the distribution of genes and establish the genotype-phenotype correlations, focusing on pediatric-onset cases.
Methods: Exome sequencing and other analytical techniques were employed to identify pathogenic variants, including duplication analysis of the PMP22 gene. Each patient underwent physical examination and electrophysiological studies. Genotypes were correlated with phenotypic features, such as age at disease onset and ulnar motor nerve conduction velocity.
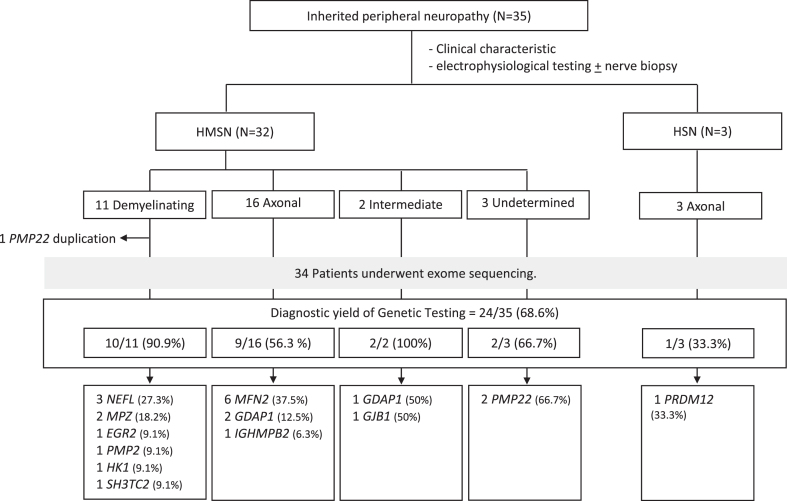
Results: We identified 35 patients with pediatric-onset inherited peripheral neuropathy. Pathogenic or likely pathogenic variants were confirmed in 24 out of 35 (68.6%) patients, with 4 of these variants being novel. A confirmed molecular diagnosis was achieved in 90.9% (10/11) of patients with demyelinating Charcot-Marie-Tooth disease (CMT) and 56.3% (9/16) of patients with axonal CMT. Among patients with infantile-onset CMT (≤2 years), the most common causative genes were MFN2 and NEFL, while GDAP1 and MFN2 were frequent causes among patients with childhood- or adolescent-onset CMT (3-9 years).
Conclusions: The MFN2 gene was the most commonly implicated gene, and the axonal type was predominant in this cohort of Thai patients with pediatric-onset inherited peripheral neuropathy.
期刊介绍:
The Journal of Neuromuscular Diseases aims to facilitate progress in understanding the molecular genetics/correlates, pathogenesis, pharmacology, diagnosis and treatment of acquired and genetic neuromuscular diseases (including muscular dystrophy, myasthenia gravis, spinal muscular atrophy, neuropathies, myopathies, myotonias and myositis). The journal publishes research reports, reviews, short communications, letters-to-the-editor, and will consider research that has negative findings. The journal is dedicated to providing an open forum for original research in basic science, translational and clinical research that will improve our fundamental understanding and lead to effective treatments of neuromuscular diseases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
