Betty Y.T. Lee, Andrew D. Phillips*, Muhammad Hanif, Tilo Söhnel and Christian G. Hartinger*,
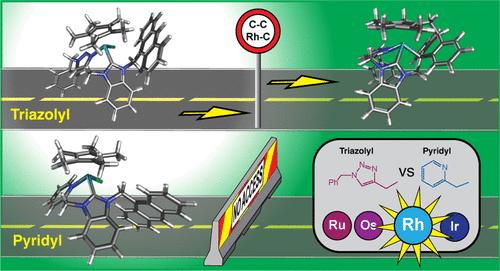
{"title":"Triazolyl- vs Pyridyl-Functionalized N-Heterocyclic Carbene Complexes: Impact of the Pendant N-Donor Ligand on Intramolecular C–C Bond Formation","authors":"Betty Y.T. Lee, Andrew D. Phillips*, Muhammad Hanif, Tilo Söhnel and Christian G. Hartinger*, ","doi":"10.1021/acsorginorgau.2c00035","DOIUrl":null,"url":null,"abstract":"<p >Organometallic Rh(Cp*) (Cp* = η<sup>5</sup>-pentamethylcyclopentadienyl) complexes with monodentate <i>N</i>-heterocyclic carbene (NHC) ligands bearing a pendant anthracenyl substituent have been shown to undergo intramolecular C–C coupling reactions. Herein, two bidentate NHC ligands substituted with pyridyl or triazolyl donor groups were prepared along with the corresponding M<sup>II/III</sup> (M = Ru<sup>II</sup>, Os<sup>II</sup>, Rh<sup>III</sup>, Ir<sup>III</sup>) complexes. While the Rh(Cp*) complex featuring an NHC-triazole bidentate ligand underwent the equivalent reaction as the monodentate Rh(NHC) complex, <i>i.e.</i>, it formed a polydentate ligand, the pyridyl-pendant derivative was unequivocally shown to be unreactive. This contrasting behavior was further investigated by density functional theory (DFT) calculations that highlighted significant differences between the two types of Rh(III) complexes with pendant pyridyl or triazolyl <i>N</i>-coordinating groups. Modeling of the reaction pathways suggests that the initial formation of a dicationic Rh(III) species is unfavorable and that the internal ligand transformation proceeds first by dissociation of the coordinated N atom of the pendant group from the Rh center. After the formation of a neutral η<sup>4</sup>-fulvene ligand <i>via</i> combined proton/single electron transfer, a cycloaddition occurs between the exo-ene bond of fulvene and the 9′ and 10′ positions on the pendant anthracenyl group. The resulting experimental UV–visible spectrum recorded in methanol of the polydentate triazolyl-based Rh species revealed the loss of the vibronic coupling typically associated with an anthracenyl functional group. Moreover, TD-DFT modeling indicates the presence of an equilibrium process whereby the <i>N</i>-coordination of the pendant triazolyl group to the Rh<sup>III</sup> center appears to be highly labile. Charge decomposition analysis (CDA) of the DFT-modeled species with the dissociated triazolyl group revealed a pseudo-η<sup>3</sup>-allylic interaction between the π-type MOs of the transformed anthracenyl group and the Rh<sup>III</sup> center; thus, the singly attached chelating ligand is classified as having rare nonadenticity.</p>","PeriodicalId":29797,"journal":{"name":"ACS Organic & Inorganic Au","volume":"2 6","pages":"511–524"},"PeriodicalIF":3.3000,"publicationDate":"2022-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acsorginorgau.2c00035","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Organic & Inorganic Au","FirstCategoryId":"1085","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acsorginorgau.2c00035","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Organometallic Rh(Cp*) (Cp* = η5-pentamethylcyclopentadienyl) complexes with monodentate N-heterocyclic carbene (NHC) ligands bearing a pendant anthracenyl substituent have been shown to undergo intramolecular C–C coupling reactions. Herein, two bidentate NHC ligands substituted with pyridyl or triazolyl donor groups were prepared along with the corresponding MII/III (M = RuII, OsII, RhIII, IrIII) complexes. While the Rh(Cp*) complex featuring an NHC-triazole bidentate ligand underwent the equivalent reaction as the monodentate Rh(NHC) complex, i.e., it formed a polydentate ligand, the pyridyl-pendant derivative was unequivocally shown to be unreactive. This contrasting behavior was further investigated by density functional theory (DFT) calculations that highlighted significant differences between the two types of Rh(III) complexes with pendant pyridyl or triazolyl N-coordinating groups. Modeling of the reaction pathways suggests that the initial formation of a dicationic Rh(III) species is unfavorable and that the internal ligand transformation proceeds first by dissociation of the coordinated N atom of the pendant group from the Rh center. After the formation of a neutral η4-fulvene ligand via combined proton/single electron transfer, a cycloaddition occurs between the exo-ene bond of fulvene and the 9′ and 10′ positions on the pendant anthracenyl group. The resulting experimental UV–visible spectrum recorded in methanol of the polydentate triazolyl-based Rh species revealed the loss of the vibronic coupling typically associated with an anthracenyl functional group. Moreover, TD-DFT modeling indicates the presence of an equilibrium process whereby the N-coordination of the pendant triazolyl group to the RhIII center appears to be highly labile. Charge decomposition analysis (CDA) of the DFT-modeled species with the dissociated triazolyl group revealed a pseudo-η3-allylic interaction between the π-type MOs of the transformed anthracenyl group and the RhIII center; thus, the singly attached chelating ligand is classified as having rare nonadenticity.
期刊介绍:
ACS Organic & Inorganic Au is an open access journal that publishes original experimental and theoretical/computational studies on organic organometallic inorganic crystal growth and engineering and organic process chemistry. Short letters comprehensive articles reviews and perspectives are welcome on topics that include:Organic chemistry Organometallic chemistry Inorganic Chemistry and Organic Process Chemistry.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
