{"title":"A review on computational modeling of instability and degradation issues of halide perovskite photovoltaic materials","authors":"Pranjul Bhatt, Ayush Kumar Pandey, Ashutosh Rajput, Kshitij Kumar Sharma, Abdul Moyez, Abhishek Tewari","doi":"10.1002/wcms.1677","DOIUrl":null,"url":null,"abstract":"<p>Hybrid halide perovskite solar cells have been recognized as one of the most promising future photovoltaic technologies due to their demonstrated high-power conversion efficiency, versatile stoichiometry and low cost. However, degradation caused by environmental exposure and structural instability due to ionic defect migration hinders the commercialization of this technology. While the experimental studies try to understand the phenomenology of the degradation mechanisms and devise practical measures to improve the stability of these materials, theoretical studies have attempted to bridge the gaps in our understanding of the fundamental degradation mechanisms at different time and length scales. A deeper understanding of the physical and chemical phenomena at an atomic level through multiscale materials modeling is going to be crucial for the knowledge-based prognosis and design of future halide perovskites. There have been increased efforts in this direction in the last few years. However, the instability fundamentals explored through atomistic modeling and simulation methods have not been reviewed comprehensively in the literature yet. Therefore, this paper is an attempt to present a critical review, while identifying the existing gaps and opportunities in the investigation of the degradation and instability issues of the halide perovskites using computational methods. The review will primarily focus on the instability caused due to the intrinsic ionic defect migration and degradation due to thermal, moisture and light exposure. The findings from the simulation studies conducted primarily using density functional theory, ab initio molecular dynamics, classical molecular dynamics and machine learning methods will be presented.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"13 6","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2023-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/wcms.1677","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 1
Abstract
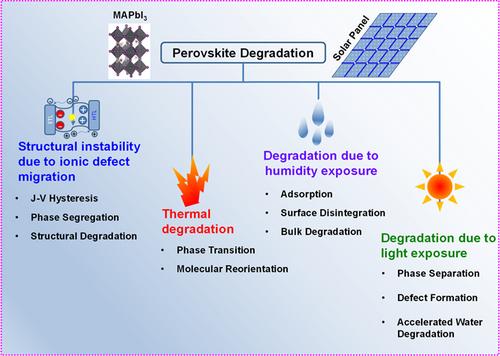
Hybrid halide perovskite solar cells have been recognized as one of the most promising future photovoltaic technologies due to their demonstrated high-power conversion efficiency, versatile stoichiometry and low cost. However, degradation caused by environmental exposure and structural instability due to ionic defect migration hinders the commercialization of this technology. While the experimental studies try to understand the phenomenology of the degradation mechanisms and devise practical measures to improve the stability of these materials, theoretical studies have attempted to bridge the gaps in our understanding of the fundamental degradation mechanisms at different time and length scales. A deeper understanding of the physical and chemical phenomena at an atomic level through multiscale materials modeling is going to be crucial for the knowledge-based prognosis and design of future halide perovskites. There have been increased efforts in this direction in the last few years. However, the instability fundamentals explored through atomistic modeling and simulation methods have not been reviewed comprehensively in the literature yet. Therefore, this paper is an attempt to present a critical review, while identifying the existing gaps and opportunities in the investigation of the degradation and instability issues of the halide perovskites using computational methods. The review will primarily focus on the instability caused due to the intrinsic ionic defect migration and degradation due to thermal, moisture and light exposure. The findings from the simulation studies conducted primarily using density functional theory, ab initio molecular dynamics, classical molecular dynamics and machine learning methods will be presented.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
