Min Liu, Lu Zhang, Xinyi Qin, Tao Huang, Ziwei Xu, Guangzhong Liu
{"title":"Prediction of Nitration Sites Based on FCBF Method and Stacking Ensemble Model","authors":"Min Liu, Lu Zhang, Xinyi Qin, Tao Huang, Ziwei Xu, Guangzhong Liu","doi":"10.2174/1570164618999210101222637","DOIUrl":null,"url":null,"abstract":"Nitration is one of the important Post-Translational Modification (PTM) occurring on the tyrosine residues of proteins. The occurrence of protein tyrosine nitration under disease conditions is inevitable and represents a shift from the signal transducing physiological actions of -NO to oxidative and potentially pathogenic pathways. Abnormal protein nitration modification can lead to serious human diseases, including neurodegenerative diseases, acute respiratory distress, organ transplant rejection and lung cancer. It is necessary and important to identify the nitration sites in protein sequences. Predicting that which tyrosine residues in the protein sequence are nitrated and which are not is of great significance for the study of nitration mechanism and related diseases. In this study, a prediction model of nitration sites based on the over-under sampling strategy and the FCBF method was proposed by stacking ensemble learning and fusing multiple features. Firstly, the protein sequence sample was encoded by 2701-dimensional fusion features (PseAAC, PSSM, AAIndex, CKSAAP, Disorder). Secondly, the ranked feature set was generated by the FCBF method according to the symmetric uncertainty metric. Thirdly, in the process of model training, use the over- and under- sampling technique was used to tackle the imbalanced dataset. Finally, the Incremental Feature Selection (IFS) method was adopted to extract an optimal classifier based on 10-fold cross-validation. Results show that the model has significant performance advantages in indicators such as MCC, Recall and F1-score, no matter in what way the comparison was conducted with other classifiers on the independent test set, or made by cross-validation with single-type feature or with fusion-features on the training set. By integrating the FCBF feature ranking methods, over- and under- sampling technique and a stacking model composed of multiple base classifiers, an effective prediction model for nitration PTM sites was build, which can achieve a better recall rate when the ratio of positive and negative samples is highly imbalanced.","PeriodicalId":50601,"journal":{"name":"Current Proteomics","volume":"29 1","pages":"1-11"},"PeriodicalIF":0.5000,"publicationDate":"2021-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Proteomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.2174/1570164618999210101222637","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 3
Abstract
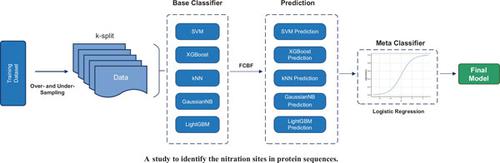
Nitration is one of the important Post-Translational Modification (PTM) occurring on the tyrosine residues of proteins. The occurrence of protein tyrosine nitration under disease conditions is inevitable and represents a shift from the signal transducing physiological actions of -NO to oxidative and potentially pathogenic pathways. Abnormal protein nitration modification can lead to serious human diseases, including neurodegenerative diseases, acute respiratory distress, organ transplant rejection and lung cancer. It is necessary and important to identify the nitration sites in protein sequences. Predicting that which tyrosine residues in the protein sequence are nitrated and which are not is of great significance for the study of nitration mechanism and related diseases. In this study, a prediction model of nitration sites based on the over-under sampling strategy and the FCBF method was proposed by stacking ensemble learning and fusing multiple features. Firstly, the protein sequence sample was encoded by 2701-dimensional fusion features (PseAAC, PSSM, AAIndex, CKSAAP, Disorder). Secondly, the ranked feature set was generated by the FCBF method according to the symmetric uncertainty metric. Thirdly, in the process of model training, use the over- and under- sampling technique was used to tackle the imbalanced dataset. Finally, the Incremental Feature Selection (IFS) method was adopted to extract an optimal classifier based on 10-fold cross-validation. Results show that the model has significant performance advantages in indicators such as MCC, Recall and F1-score, no matter in what way the comparison was conducted with other classifiers on the independent test set, or made by cross-validation with single-type feature or with fusion-features on the training set. By integrating the FCBF feature ranking methods, over- and under- sampling technique and a stacking model composed of multiple base classifiers, an effective prediction model for nitration PTM sites was build, which can achieve a better recall rate when the ratio of positive and negative samples is highly imbalanced.
Current ProteomicsBIOCHEMICAL RESEARCH METHODS-BIOCHEMISTRY & MOLECULAR BIOLOGY
CiteScore
1.60
自引率
0.00%
发文量
25
审稿时长
>0 weeks
期刊介绍:
Research in the emerging field of proteomics is growing at an extremely rapid rate. The principal aim of Current Proteomics is to publish well-timed in-depth/mini review articles in this fast-expanding area on topics relevant and significant to the development of proteomics. Current Proteomics is an essential journal for everyone involved in proteomics and related fields in both academia and industry.
Current Proteomics publishes in-depth/mini review articles in all aspects of the fast-expanding field of proteomics. All areas of proteomics are covered together with the methodology, software, databases, technological advances and applications of proteomics, including functional proteomics. Diverse technologies covered include but are not limited to:
Protein separation and characterization techniques
2-D gel electrophoresis and image analysis
Techniques for protein expression profiling including mass spectrometry-based methods and algorithms for correlative database searching
Determination of co-translational and post- translational modification of proteins
Protein/peptide microarrays
Biomolecular interaction analysis
Analysis of protein complexes
Yeast two-hybrid projects
Protein-protein interaction (protein interactome) pathways and cell signaling networks
Systems biology
Proteome informatics (bioinformatics)
Knowledge integration and management tools
High-throughput protein structural studies (using mass spectrometry, nuclear magnetic resonance and X-ray crystallography)
High-throughput computational methods for protein 3-D structure as well as function determination
Robotics, nanotechnology, and microfluidics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
