Maeson S Latsko, Daniel C Koboldt, Samuel J Franklin, Scott E Hickey, Rachel K Williamson, Shannon Garner, Adam P Ostendorf, Kristy Lee, Peter White, Richard K Wilson
{"title":"De novo missense mutation in GRIA2 in a patient with global developmental delay, autism spectrum disorder, and epileptic encephalopathy.","authors":"Maeson S Latsko, Daniel C Koboldt, Samuel J Franklin, Scott E Hickey, Rachel K Williamson, Shannon Garner, Adam P Ostendorf, Kristy Lee, Peter White, Richard K Wilson","doi":"10.1101/mcs.a006172","DOIUrl":null,"url":null,"abstract":"<p><p>De novo variants are increasingly recognized as a common cause of early infantile epileptic encephalopathies. We present a 4-year-old male with epileptic encephalopathy characterized by seizures, autism spectrum disorder, and global developmental delay. Whole genome sequencing of the proband and his unaffected parents revealed a novel de novo missense variant in GRIA2 (c.1589A>T; p.Lys530Met; ENST00000264426.14). Variants in the GRIA2 gene were recently reported to cause an autosomal dominant neurodevelopmental disorder with language impairments and behavioral abnormalities (OMIM; MIM #618917), a condition characterized by intellectual disability and developmental delay in which seizures are a common feature. The de novo variant identified in our patient maps to the edge of a key ligand binding domain of the AMPA receptor and has not been previously reported in gnomAD or other public databases, making it novel. Our findings provided a long-sought diagnosis for this patient and support the link between GRIA2 and a dominant neurodevelopmental disorder.</p>","PeriodicalId":50995,"journal":{"name":"Coastal Management","volume":"45 1","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2022-05-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9235849/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Coastal Management","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006172","RegionNum":4,"RegionCategory":"环境科学与生态学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ENVIRONMENTAL SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
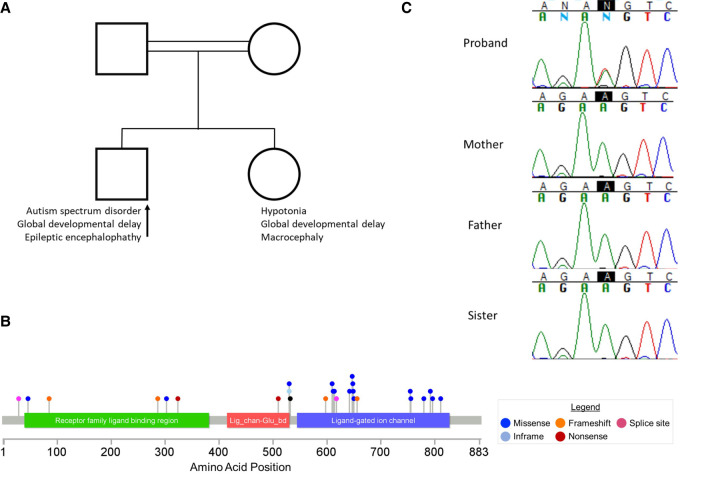
De novo variants are increasingly recognized as a common cause of early infantile epileptic encephalopathies. We present a 4-year-old male with epileptic encephalopathy characterized by seizures, autism spectrum disorder, and global developmental delay. Whole genome sequencing of the proband and his unaffected parents revealed a novel de novo missense variant in GRIA2 (c.1589A>T; p.Lys530Met; ENST00000264426.14). Variants in the GRIA2 gene were recently reported to cause an autosomal dominant neurodevelopmental disorder with language impairments and behavioral abnormalities (OMIM; MIM #618917), a condition characterized by intellectual disability and developmental delay in which seizures are a common feature. The de novo variant identified in our patient maps to the edge of a key ligand binding domain of the AMPA receptor and has not been previously reported in gnomAD or other public databases, making it novel. Our findings provided a long-sought diagnosis for this patient and support the link between GRIA2 and a dominant neurodevelopmental disorder.
期刊介绍:
Coastal Management is an international peer-reviewed, applied research journal dedicated to exploring the technical, applied ecological, legal, political, social, and policy issues relating to the use of coastal and ocean resources and environments on a global scale. The journal presents timely information on management tools and techniques as well as recent findings from research and analysis that bear directly on management and policy. Findings must be grounded in the current peer reviewed literature and relevant studies. Articles must contain a clear and relevant management component. Preference is given to studies of interest to an international readership, but case studies are accepted if conclusions are derived from acceptable evaluative methods, reference to comparable cases, and related to peer reviewed studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
