{"title":"Genetic Links to Episodic Movement Disorders: Current Insights.","authors":"Divyani Garg, Shekeeb Mohammad, Anju Shukla, Suvasini Sharma","doi":"10.2147/TACG.S363485","DOIUrl":null,"url":null,"abstract":"<p><p>Episodic or paroxysmal movement disorders (PxMD) are conditions, which occur episodically, are transient, usually have normal interictal periods, and are characterized by hyperkinetic disorders, including ataxia, chorea, dystonia, and ballism. Broadly, these comprise paroxysmal dyskinesias (paroxysmal kinesigenic and non-kinesigenic dyskinesia [PKD/PNKD], paroxysmal exercise-induced dyskinesias [PED]) and episodic ataxias (EA) types 1-9. Classification of paroxysmal dyskinesias has traditionally been clinical. However, with advancement in genetics and the discovery of the molecular basis of several of these disorders, it is becoming clear that phenotypic pleiotropy exists, that is, the same variant may give rise to a variety of phenotypes, and the classical understanding of these disorders requires a new paradigm. Based on molecular pathogenesis, paroxysmal disorders are now categorized as synaptopathies, transportopathies, channelopathies, second-messenger related disorders, mitochondrial or others. A genetic paradigm also has an advantage of identifying potentially treatable disorders, such as glucose transporter 1 deficiency syndromes, which necessitates a ketogenic diet, and <i>ADCY5</i>-related disorders, which may respond to caffeine. Clues for a primary etiology include age at onset below 18 years, presence of family history and fixed triggers and attack duration. Paroxysmal movement disorder is a network disorder, with both the basal ganglia and the cerebellum implicated in pathogenesis. Abnormalities in the striatal cAMP turnover pathway may also be contributory. Although next-generation sequencing has restructured the approach to paroxysmal movement disorders, the genetic underpinnings of several entities remain undiscovered. As more genes and variants continue to be reported, these will lead to enhanced understanding of pathophysiological mechanisms and precise treatment.</p>","PeriodicalId":74945,"journal":{"name":"","volume":"16 ","pages":"11-30"},"PeriodicalIF":0.0,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/5b/99/tacg-16-11.PMC9985884.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S363485","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
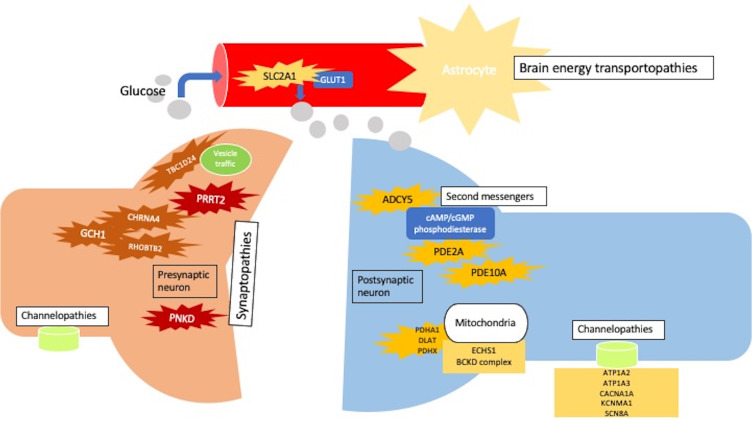
Abstract
Episodic or paroxysmal movement disorders (PxMD) are conditions, which occur episodically, are transient, usually have normal interictal periods, and are characterized by hyperkinetic disorders, including ataxia, chorea, dystonia, and ballism. Broadly, these comprise paroxysmal dyskinesias (paroxysmal kinesigenic and non-kinesigenic dyskinesia [PKD/PNKD], paroxysmal exercise-induced dyskinesias [PED]) and episodic ataxias (EA) types 1-9. Classification of paroxysmal dyskinesias has traditionally been clinical. However, with advancement in genetics and the discovery of the molecular basis of several of these disorders, it is becoming clear that phenotypic pleiotropy exists, that is, the same variant may give rise to a variety of phenotypes, and the classical understanding of these disorders requires a new paradigm. Based on molecular pathogenesis, paroxysmal disorders are now categorized as synaptopathies, transportopathies, channelopathies, second-messenger related disorders, mitochondrial or others. A genetic paradigm also has an advantage of identifying potentially treatable disorders, such as glucose transporter 1 deficiency syndromes, which necessitates a ketogenic diet, and ADCY5-related disorders, which may respond to caffeine. Clues for a primary etiology include age at onset below 18 years, presence of family history and fixed triggers and attack duration. Paroxysmal movement disorder is a network disorder, with both the basal ganglia and the cerebellum implicated in pathogenesis. Abnormalities in the striatal cAMP turnover pathway may also be contributory. Although next-generation sequencing has restructured the approach to paroxysmal movement disorders, the genetic underpinnings of several entities remain undiscovered. As more genes and variants continue to be reported, these will lead to enhanced understanding of pathophysiological mechanisms and precise treatment.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
