Dorothea Seiler Vellame, Gemma Shireby, Ailsa MacCalman, Emma L Dempster, Joe Burrage, Tyler Gorrie-Stone, Leonard S Schalkwyk, Jonathan Mill, Eilis Hannon
{"title":"Uncertainty quantification of reference-based cellular deconvolution algorithms.","authors":"Dorothea Seiler Vellame, Gemma Shireby, Ailsa MacCalman, Emma L Dempster, Joe Burrage, Tyler Gorrie-Stone, Leonard S Schalkwyk, Jonathan Mill, Eilis Hannon","doi":"10.1080/15592294.2022.2137659","DOIUrl":null,"url":null,"abstract":"<p><p>The majority of epigenetic epidemiology studies to date have generated genome-wide profiles from bulk tissues (e.g., whole blood) however these are vulnerable to confounding from variation in cellular composition. Proxies for cellular composition can be mathematically derived from the bulk tissue profiles using a deconvolution algorithm; however, there is no method to assess the validity of these estimates for a dataset where the true cellular proportions are unknown. In this study, we describe, validate and characterize a sample level accuracy metric for derived cellular heterogeneity variables. The CETYGO score captures the deviation between a sample's DNA methylation profile and its expected profile given the estimated cellular proportions and cell type reference profiles. We demonstrate that the CETYGO score consistently distinguishes inaccurate and incomplete deconvolutions when applied to reconstructed whole blood profiles. By applying our novel metric to >6,300 empirical whole blood profiles, we find that estimating accurate cellular composition is influenced by both technical and biological variation. In particular, we show that when using a common reference panel for whole blood, less accurate estimates are generated for females, neonates, older individuals and smokers. Our results highlight the utility of a metric to assess the accuracy of cellular deconvolution, and describe how it can enhance studies of DNA methylation that are reliant on statistical proxies for cellular heterogeneity. To facilitate incorporating our methodology into existing pipelines, we have made it freely available as an R package (https://github.com/ds420/CETYGO).</p>","PeriodicalId":11767,"journal":{"name":"Epigenetics","volume":"18 1","pages":"2137659"},"PeriodicalIF":3.2000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9980651/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Epigenetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1080/15592294.2022.2137659","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/12/20 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
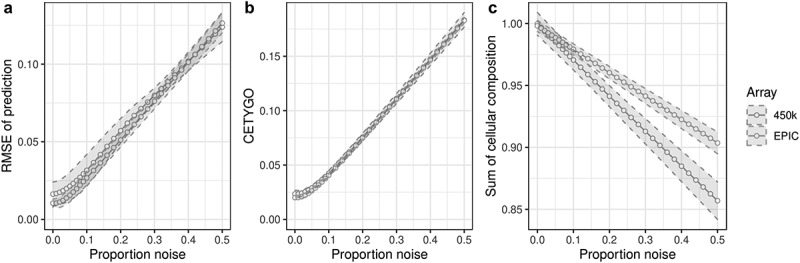
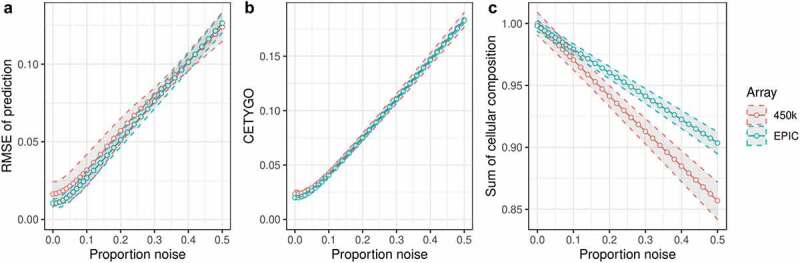
The majority of epigenetic epidemiology studies to date have generated genome-wide profiles from bulk tissues (e.g., whole blood) however these are vulnerable to confounding from variation in cellular composition. Proxies for cellular composition can be mathematically derived from the bulk tissue profiles using a deconvolution algorithm; however, there is no method to assess the validity of these estimates for a dataset where the true cellular proportions are unknown. In this study, we describe, validate and characterize a sample level accuracy metric for derived cellular heterogeneity variables. The CETYGO score captures the deviation between a sample's DNA methylation profile and its expected profile given the estimated cellular proportions and cell type reference profiles. We demonstrate that the CETYGO score consistently distinguishes inaccurate and incomplete deconvolutions when applied to reconstructed whole blood profiles. By applying our novel metric to >6,300 empirical whole blood profiles, we find that estimating accurate cellular composition is influenced by both technical and biological variation. In particular, we show that when using a common reference panel for whole blood, less accurate estimates are generated for females, neonates, older individuals and smokers. Our results highlight the utility of a metric to assess the accuracy of cellular deconvolution, and describe how it can enhance studies of DNA methylation that are reliant on statistical proxies for cellular heterogeneity. To facilitate incorporating our methodology into existing pipelines, we have made it freely available as an R package (https://github.com/ds420/CETYGO).
期刊介绍:
Epigenetics publishes peer-reviewed original research and review articles that provide an unprecedented forum where epigenetic mechanisms and their role in diverse biological processes can be revealed, shared, and discussed.
Epigenetics research studies heritable changes in gene expression caused by mechanisms others than the modification of the DNA sequence. Epigenetics therefore plays critical roles in a variety of biological systems, diseases, and disciplines. Topics of interest include (but are not limited to):
DNA methylation
Nucleosome positioning and modification
Gene silencing
Imprinting
Nuclear reprogramming
Chromatin remodeling
Non-coding RNA
Non-histone chromosomal elements
Dosage compensation
Nuclear organization
Epigenetic therapy and diagnostics
Nutrition and environmental epigenetics
Cancer epigenetics
Neuroepigenetics




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
