Abdulrahman Alasiri, Konrad J Karczewski, Brian Cole, Bao-Li Loza, Jason H Moore, Sander W van der Laan, Folkert W Asselbergs, Brendan J Keating, Jessica van Setten
{"title":"LoFTK: a framework for fully automated calculation of predicted Loss-of-Function variants and genes.","authors":"Abdulrahman Alasiri, Konrad J Karczewski, Brian Cole, Bao-Li Loza, Jason H Moore, Sander W van der Laan, Folkert W Asselbergs, Brendan J Keating, Jessica van Setten","doi":"10.1186/s13040-023-00321-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Loss-of-Function (LoF) variants in human genes are important due to their impact on clinical phenotypes and frequent occurrence in the genomes of healthy individuals. The association of LoF variants with complex diseases and traits may lead to the discovery and validation of novel therapeutic targets. Current approaches predict high-confidence LoF variants without identifying the specific genes or the number of copies they affect. Moreover, there is a lack of methods for detecting knockout genes caused by compound heterozygous (CH) LoF variants.</p><p><strong>Results: </strong>We have developed the Loss-of-Function ToolKit (LoFTK), which allows efficient and automated prediction of LoF variants from genotyped, imputed and sequenced genomes. LoFTK enables the identification of genes that are inactive in one or two copies and provides summary statistics for downstream analyses. LoFTK can identify CH LoF variants, which result in LoF genes with two copies lost. Using data from parents and offspring we show that 96% of CH LoF genes predicted by LoFTK in the offspring have the respective alleles donated by each parent.</p><p><strong>Conclusions: </strong>LoFTK is a command-line based tool that provides a reliable computational workflow for predicting LoF variants from genotyped and sequenced genomes, identifying genes that are inactive in 1 or 2 copies. LoFTK is an open software and is freely available to non-commercial users at https://github.com/CirculatoryHealth/LoFTK .</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"16 1","pages":"3"},"PeriodicalIF":6.1000,"publicationDate":"2023-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9893534/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-023-00321-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Loss-of-Function (LoF) variants in human genes are important due to their impact on clinical phenotypes and frequent occurrence in the genomes of healthy individuals. The association of LoF variants with complex diseases and traits may lead to the discovery and validation of novel therapeutic targets. Current approaches predict high-confidence LoF variants without identifying the specific genes or the number of copies they affect. Moreover, there is a lack of methods for detecting knockout genes caused by compound heterozygous (CH) LoF variants.
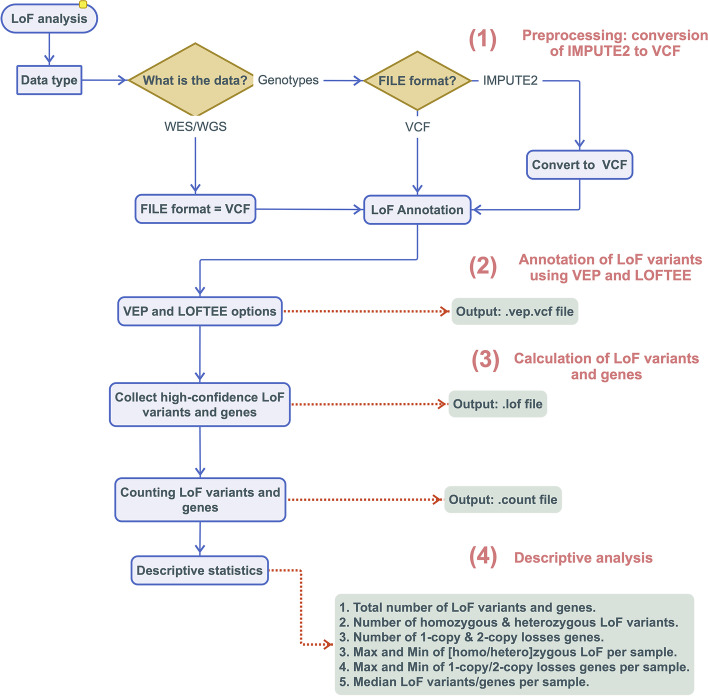
Results: We have developed the Loss-of-Function ToolKit (LoFTK), which allows efficient and automated prediction of LoF variants from genotyped, imputed and sequenced genomes. LoFTK enables the identification of genes that are inactive in one or two copies and provides summary statistics for downstream analyses. LoFTK can identify CH LoF variants, which result in LoF genes with two copies lost. Using data from parents and offspring we show that 96% of CH LoF genes predicted by LoFTK in the offspring have the respective alleles donated by each parent.
Conclusions: LoFTK is a command-line based tool that provides a reliable computational workflow for predicting LoF variants from genotyped and sequenced genomes, identifying genes that are inactive in 1 or 2 copies. LoFTK is an open software and is freely available to non-commercial users at https://github.com/CirculatoryHealth/LoFTK .
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
