Mariana Sant'Anna, Elisa Gravito-Soares, Marta Gravito-Soares, Sofia Mendes, Pedro Narra Figueiredo
{"title":"Solitary Peutz-Jeghers Type Hamartomatous Polyp Arising from the Appendix.","authors":"Mariana Sant'Anna, Elisa Gravito-Soares, Marta Gravito-Soares, Sofia Mendes, Pedro Narra Figueiredo","doi":"10.1159/000521196","DOIUrl":null,"url":null,"abstract":"Digestive hamartomatous polyps may be solitary or multiple, the latter often associated with genetic predisposition [1, 2]. Solitary Peutz-Jeghers (PJ)-type hamartomatous polyps represent a rare and distinct entity from the classic PJ syndrome, an autosomal dominant genetic disorder characterized by the development of multiple polyps in the gastrointestinal (GI) tract in association with patches of hyperpigmentation in the mouth, hands and feet [1, 3]. It is important to distinguish these two entities since the latter is associated with a lifetime cumulative risk of up to 93% for development of malignancies (such as colorectal, breast, small bowel, gastric and pancreatic cancers), but the first seems benign in its course. Solitary PJ polyps are diagnosed in patients with an isolated hamartomatous polyp of the GI tract, no familiar history of polyposis and no typical phenotype [3]. An 80-year-old man with a history of sigmoidectomy due to an obstructive T2N0M0 colorectal cancer underwent a surveillance thoracoabdominopelvic CT scan showing a voluminous endoluminal polyp at the caecum (Fig. 1). He was referred to our reference centre for colonoscopy. A 4-cm diameter pedunculated and congestive polyp was identified with a short and thick stalk arising from the appendiceal orifice. We proceeded to its en bloc resection using a 25-mm oval diathermic snare after stalk injection with diluted adrenaline 1:10,000, normal saline and methylene blue with polyp recovery for histology (Fig. 2a–c, 3a). The polyp was confirmed to be a hamartomatous polyp of the PJ type (R0 resection) with arborizing pattern of vascularized smooth-muscle tissue axes covered by elongated veliform crypts and occasional intraluminal necrosis with no dysplasia (Fig. 3b). The patient had no typical manifestations of PJ syndrome or family history. His follow-up showed no complications related to the procedure, including bleeding or acute appendicitis (prophylactic antibiotic was not used). Solitary PJ-type hamartomatous polyps of the GI tract are rare, and appendiceal location is even rarer. Accord-","PeriodicalId":51838,"journal":{"name":"GE Portuguese Journal of Gastroenterology","volume":"30 2","pages":"156-158"},"PeriodicalIF":0.6000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/84/72/pjg-0030-0156.PMC10050839.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"GE Portuguese Journal of Gastroenterology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000521196","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
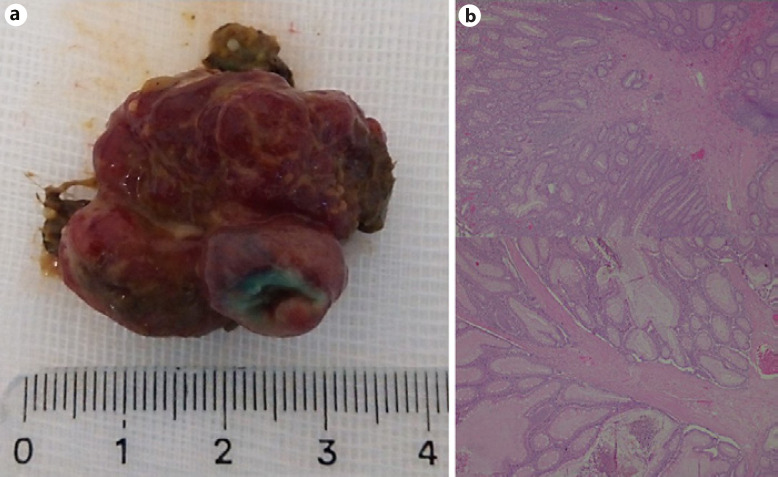
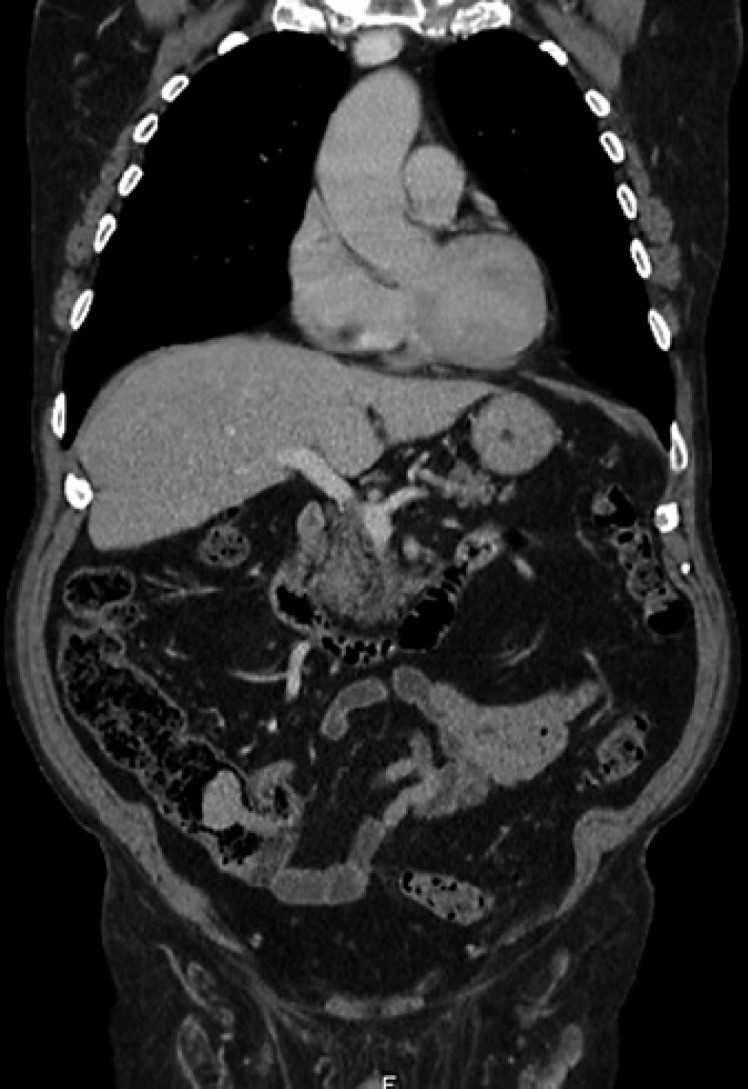
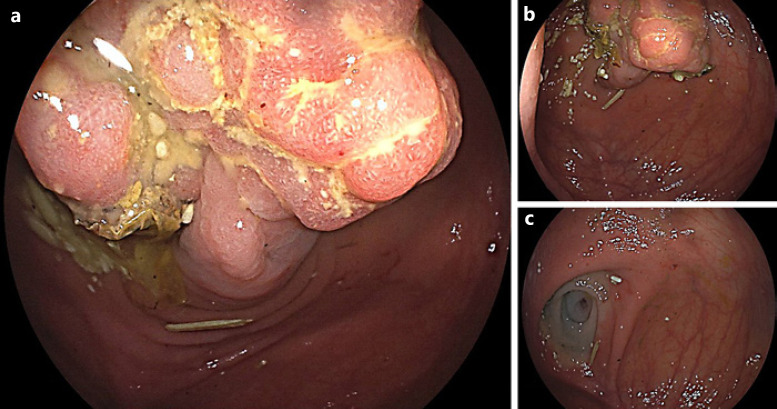
Digestive hamartomatous polyps may be solitary or multiple, the latter often associated with genetic predisposition [1, 2]. Solitary Peutz-Jeghers (PJ)-type hamartomatous polyps represent a rare and distinct entity from the classic PJ syndrome, an autosomal dominant genetic disorder characterized by the development of multiple polyps in the gastrointestinal (GI) tract in association with patches of hyperpigmentation in the mouth, hands and feet [1, 3]. It is important to distinguish these two entities since the latter is associated with a lifetime cumulative risk of up to 93% for development of malignancies (such as colorectal, breast, small bowel, gastric and pancreatic cancers), but the first seems benign in its course. Solitary PJ polyps are diagnosed in patients with an isolated hamartomatous polyp of the GI tract, no familiar history of polyposis and no typical phenotype [3]. An 80-year-old man with a history of sigmoidectomy due to an obstructive T2N0M0 colorectal cancer underwent a surveillance thoracoabdominopelvic CT scan showing a voluminous endoluminal polyp at the caecum (Fig. 1). He was referred to our reference centre for colonoscopy. A 4-cm diameter pedunculated and congestive polyp was identified with a short and thick stalk arising from the appendiceal orifice. We proceeded to its en bloc resection using a 25-mm oval diathermic snare after stalk injection with diluted adrenaline 1:10,000, normal saline and methylene blue with polyp recovery for histology (Fig. 2a–c, 3a). The polyp was confirmed to be a hamartomatous polyp of the PJ type (R0 resection) with arborizing pattern of vascularized smooth-muscle tissue axes covered by elongated veliform crypts and occasional intraluminal necrosis with no dysplasia (Fig. 3b). The patient had no typical manifestations of PJ syndrome or family history. His follow-up showed no complications related to the procedure, including bleeding or acute appendicitis (prophylactic antibiotic was not used). Solitary PJ-type hamartomatous polyps of the GI tract are rare, and appendiceal location is even rarer. Accord-
期刊介绍:
The ''GE Portuguese Journal of Gastroenterology'' (formerly Jornal Português de Gastrenterologia), founded in 1994, is the official publication of Sociedade Portuguesa de Gastrenterologia (Portuguese Society of Gastroenterology), Sociedade Portuguesa de Endoscopia Digestiva (Portuguese Society of Digestive Endoscopy) and Associação Portuguesa para o Estudo do Fígado (Portuguese Association for the Study of the Liver). The journal publishes clinical and basic research articles on Gastroenterology, Digestive Endoscopy, Hepatology and related topics. Review articles, clinical case studies, images, letters to the editor and other articles such as recommendations or papers on gastroenterology clinical practice are also considered. Only articles written in English are accepted.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
