Yujie Xu, Haozhe Xu, Tao Ling, Yachao Cui, Junwei Zhang, Xianmin Mu, Desheng Zhou, Ting Zhao, Yingchang Li, Zhongping Su, Qiang You
{"title":"Inhibitor of nuclear factor kappa B kinase subunit epsilon regulates murine acetaminophen toxicity via RIPK1/JNK.","authors":"Yujie Xu, Haozhe Xu, Tao Ling, Yachao Cui, Junwei Zhang, Xianmin Mu, Desheng Zhou, Ting Zhao, Yingchang Li, Zhongping Su, Qiang You","doi":"10.1007/s10565-023-09796-8","DOIUrl":null,"url":null,"abstract":"<p><p>Drug-induced liver injury (DILI) still poses a major clinical challenge and is a leading cause of acute liver failure. Inhibitor of nuclear factor kappa B kinase subunit epsilon (IKBKE) is essential for inflammation and metabolic disorders. However, it is unclear how IKBKE regulates cellular damage in acetaminophen (APAP)-induced acute liver injury. Here, we found that the deficiency of IKBKE markedly aggravated APAP-induced acute liver injury by targeting RIPK1. We showed that APAP-treated IKBKE-deficient mice exhibited severer liver injury, worse mitochondrial integrity, and enhanced glutathione depletion than wild-type mice. IKBKE deficiency may directly upregulate the expression of total RIPK1 and the cleaved RIPK1, resulting in sustained JNK activation and increased translocation of RIPK1/JNK to mitochondria. Moreover, deficiency of IKBKE enhanced the expression of pro-inflammatory factors and inflammatory cell infiltration in the liver, especially neutrophils and monocytes. Inhibition of RIPK1 activity by necrostatin-1 significantly reduced APAP-induced liver damage. Thus, we have revealed a negative regulatory function of IKBKE, which acts as an RIPK1/JNK regulator to mediate APAP-induced hepatotoxicity. Targeting IKBKE/RIPK1 may serve as a potential therapeutic strategy for acute or chronic liver injury.</p>","PeriodicalId":9672,"journal":{"name":"Cell Biology and Toxicology","volume":" ","pages":"2709-2724"},"PeriodicalIF":5.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Biology and Toxicology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10565-023-09796-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/2/9 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
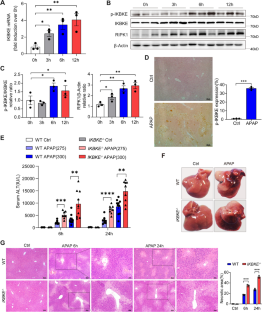
Drug-induced liver injury (DILI) still poses a major clinical challenge and is a leading cause of acute liver failure. Inhibitor of nuclear factor kappa B kinase subunit epsilon (IKBKE) is essential for inflammation and metabolic disorders. However, it is unclear how IKBKE regulates cellular damage in acetaminophen (APAP)-induced acute liver injury. Here, we found that the deficiency of IKBKE markedly aggravated APAP-induced acute liver injury by targeting RIPK1. We showed that APAP-treated IKBKE-deficient mice exhibited severer liver injury, worse mitochondrial integrity, and enhanced glutathione depletion than wild-type mice. IKBKE deficiency may directly upregulate the expression of total RIPK1 and the cleaved RIPK1, resulting in sustained JNK activation and increased translocation of RIPK1/JNK to mitochondria. Moreover, deficiency of IKBKE enhanced the expression of pro-inflammatory factors and inflammatory cell infiltration in the liver, especially neutrophils and monocytes. Inhibition of RIPK1 activity by necrostatin-1 significantly reduced APAP-induced liver damage. Thus, we have revealed a negative regulatory function of IKBKE, which acts as an RIPK1/JNK regulator to mediate APAP-induced hepatotoxicity. Targeting IKBKE/RIPK1 may serve as a potential therapeutic strategy for acute or chronic liver injury.
期刊介绍:
Cell Biology and Toxicology (CBT) is an international journal focused on clinical and translational research with an emphasis on molecular and cell biology, genetic and epigenetic heterogeneity, drug discovery and development, and molecular pharmacology and toxicology. CBT has a disease-specific scope prioritizing publications on gene and protein-based regulation, intracellular signaling pathway dysfunction, cell type-specific function, and systems in biomedicine in drug discovery and development. CBT publishes original articles with outstanding, innovative and significant findings, important reviews on recent research advances and issues of high current interest, opinion articles of leading edge science, and rapid communication or reports, on molecular mechanisms and therapies in diseases.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
