Sarah Orr, Eric Olinger, Sotia Iosifidou, Miguel Barroso-Gil, Ruxandra Neatu, Katrina Wood, Ian Wilson, Genomics England Research Consortium, John Andrew Sayer
{"title":"Molecular genetic diagnosis of kidney ciliopathies: Lessons from interpreting genomic sequencing data and the requirement for accurate phenotypic data","authors":"Sarah Orr, Eric Olinger, Sotia Iosifidou, Miguel Barroso-Gil, Ruxandra Neatu, Katrina Wood, Ian Wilson, Genomics England Research Consortium, John Andrew Sayer","doi":"10.1111/ahg.12508","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n \n <p><b>Introduction</b>: Massively parallel sequencing (MPS) techniques have made a major impact on the identification of the genetic basis of inherited kidney diseases such as the ciliopathy autosomal dominant polycystic kidney disease (ADPKD). Great care must be taken when analysing MPS data in isolation from accurate phenotypic information, as this can cause misdiagnosis. <b>Methods</b>: Here, we describe a family trio, recruited to the Genomics England 100,000 Genomes Project, labelled as having cystic kidney disease, who were genetically unsolved following routine data analysis pipelines. We performed a bespoke reanalysis of Whole Genome Sequencing (WGS) data and coupled this with revised phenotypic data and targeted PCR and Sanger sequencing to provide a precise molecular genetic diagnosis. <b>Results</b>: We detected a heterozygous <i>PKD1</i> frameshift variant within the WGS data which segregated with the redefined ADPKD phenotypes. An additional heterozygous exon deletion in <i>ALG8</i> was also found in affected and unaffected individuals, but its precise clinical significance remains unclear. <b>Conclusion</b>: This case illustrates that reanalysis of WGS data in unsolved cases of cystic kidney disease is valuable. Clinical phenotypes must be reassessed as these may have been incorrectly recorded and evolve over time. Undertaking additional studies including genotype-phenotype correlation in wider family members provides useful diagnostic information.</p>\n </section>\n </div>","PeriodicalId":8085,"journal":{"name":"Annals of Human Genetics","volume":"88 1","pages":"76-85"},"PeriodicalIF":1.2000,"publicationDate":"2023-04-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ahg.12508","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/ahg.12508","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
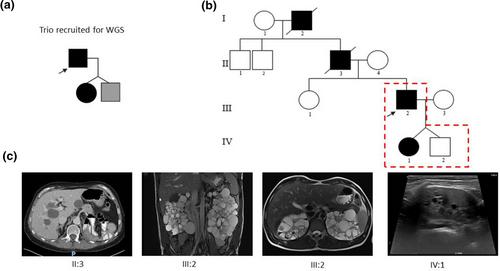
Introduction: Massively parallel sequencing (MPS) techniques have made a major impact on the identification of the genetic basis of inherited kidney diseases such as the ciliopathy autosomal dominant polycystic kidney disease (ADPKD). Great care must be taken when analysing MPS data in isolation from accurate phenotypic information, as this can cause misdiagnosis. Methods: Here, we describe a family trio, recruited to the Genomics England 100,000 Genomes Project, labelled as having cystic kidney disease, who were genetically unsolved following routine data analysis pipelines. We performed a bespoke reanalysis of Whole Genome Sequencing (WGS) data and coupled this with revised phenotypic data and targeted PCR and Sanger sequencing to provide a precise molecular genetic diagnosis. Results: We detected a heterozygous PKD1 frameshift variant within the WGS data which segregated with the redefined ADPKD phenotypes. An additional heterozygous exon deletion in ALG8 was also found in affected and unaffected individuals, but its precise clinical significance remains unclear. Conclusion: This case illustrates that reanalysis of WGS data in unsolved cases of cystic kidney disease is valuable. Clinical phenotypes must be reassessed as these may have been incorrectly recorded and evolve over time. Undertaking additional studies including genotype-phenotype correlation in wider family members provides useful diagnostic information.
期刊介绍:
Annals of Human Genetics publishes material directly concerned with human genetics or the application of scientific principles and techniques to any aspect of human inheritance. Papers that describe work on other species that may be relevant to human genetics will also be considered. Mathematical models should include examples of application to data where possible.
Authors are welcome to submit Supporting Information, such as data sets or additional figures or tables, that will not be published in the print edition of the journal, but which will be viewable via the online edition and stored on the website.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
