Laura Hecher, Frederike L Harms, Jasmin Lisfeld, Malik Alawi, Jonas Denecke, Kerstin Kutsche
{"title":"INPP4A-related genetic and phenotypic spectrum and functional relevance of subcellular targeting of INPP4A isoforms.","authors":"Laura Hecher, Frederike L Harms, Jasmin Lisfeld, Malik Alawi, Jonas Denecke, Kerstin Kutsche","doi":"10.1007/s10048-023-00709-9","DOIUrl":null,"url":null,"abstract":"<p><p>Type I inositol polyphosphate-4-phosphatase (INPP4A) belongs to the group of phosphoinositide phosphatases controlling proliferation, apoptosis, and endosome function by hydrolyzing phosphatidylinositol 3,4-bisphosphate. INPP4A produces multiple transcripts encoding shorter and longer INPP4A isoforms with hydrophilic or hydrophobic C-terminus. Biallelic INPP4A truncating variants cause a spectrum of neurodevelopmental disorders ranging from moderate intellectual disability to postnatal microcephaly with developmental and epileptic encephalopathy and (ponto)cerebellar hypoplasia. We report a girl with the novel homozygous INPP4A variant NM_001134224.2:c.2840del/p.(Gly947Glufs*12) (isoform d). She presented with postnatal microcephaly, global developmental delay, visual impairment, myoclonic seizures, and pontocerebellar hypoplasia and died at the age of 27 months. The level of mutant INPP4A mRNAs in proband-derived leukocytes was comparable to controls suggesting production of C-terminally altered INPP4A isoforms. We transiently expressed eGFP-tagged INPP4A isoform a (NM_004027.3) wildtype and p.(Gly908Glufs*12) mutant [p.(Gly947Glufs*12) according to NM_001134224.2] as well as INPP4A isoform b (NM_001566.2) wildtype and p.(Asp915Alafs*2) mutant, previously reported in family members with moderate intellectual disability, in HeLa cells and determined their subcellular distributions. While INPP4A isoform a was preferentially found in perinuclear clusters co-localizing with the GTPase Rab5, isoform b showed a net-like distribution, possibly localizing near and/or on microtubules. Quantification of intracellular localization patterns of the two INPP4A mutants revealed significant differences compared with the respective wildtype and similarity with each other. Our data suggests an important non-redundant function of INPP4A isoforms with hydrophobic or hydrophilic C-terminus in the brain.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 2","pages":"79-93"},"PeriodicalIF":1.2000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00709-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
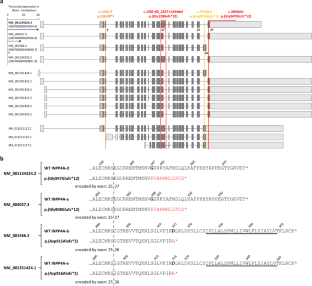
Type I inositol polyphosphate-4-phosphatase (INPP4A) belongs to the group of phosphoinositide phosphatases controlling proliferation, apoptosis, and endosome function by hydrolyzing phosphatidylinositol 3,4-bisphosphate. INPP4A produces multiple transcripts encoding shorter and longer INPP4A isoforms with hydrophilic or hydrophobic C-terminus. Biallelic INPP4A truncating variants cause a spectrum of neurodevelopmental disorders ranging from moderate intellectual disability to postnatal microcephaly with developmental and epileptic encephalopathy and (ponto)cerebellar hypoplasia. We report a girl with the novel homozygous INPP4A variant NM_001134224.2:c.2840del/p.(Gly947Glufs*12) (isoform d). She presented with postnatal microcephaly, global developmental delay, visual impairment, myoclonic seizures, and pontocerebellar hypoplasia and died at the age of 27 months. The level of mutant INPP4A mRNAs in proband-derived leukocytes was comparable to controls suggesting production of C-terminally altered INPP4A isoforms. We transiently expressed eGFP-tagged INPP4A isoform a (NM_004027.3) wildtype and p.(Gly908Glufs*12) mutant [p.(Gly947Glufs*12) according to NM_001134224.2] as well as INPP4A isoform b (NM_001566.2) wildtype and p.(Asp915Alafs*2) mutant, previously reported in family members with moderate intellectual disability, in HeLa cells and determined their subcellular distributions. While INPP4A isoform a was preferentially found in perinuclear clusters co-localizing with the GTPase Rab5, isoform b showed a net-like distribution, possibly localizing near and/or on microtubules. Quantification of intracellular localization patterns of the two INPP4A mutants revealed significant differences compared with the respective wildtype and similarity with each other. Our data suggests an important non-redundant function of INPP4A isoforms with hydrophobic or hydrophilic C-terminus in the brain.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
