Chang Li, Ian Hou, Mingjia Ma, Grace Wang, Yongsheng Bai, Xiaoming Liu
{"title":"Orthogonal analysis of variants in APOE gene using <i>in-silico</i> approaches reveals novel disrupting variants.","authors":"Chang Li, Ian Hou, Mingjia Ma, Grace Wang, Yongsheng Bai, Xiaoming Liu","doi":"10.3389/fbinf.2023.1122559","DOIUrl":null,"url":null,"abstract":"<p><p><b>Introduction:</b> Alzheimer's disease (AD) is one of the most prominent medical conditions in the world. Understanding the genetic component of the disease can greatly advance our knowledge regarding its progression, treatment and prognosis. Single amino-acid variants (SAVs) in the APOE gene have been widely investigated as a risk factor for AD Studies, including genome-wide association studies, meta-analysis based studies, and <i>in-vivo</i> animal studies, were carried out to investigate the functional importance and pathogenesis potential of APOE SAVs. However, given the high cost of such large-scale or experimental studies, there are only a handful of variants being reported that have definite explanations. The recent development of <i>in-silico</i> analytical approaches, especially large-scale deep learning models, has opened new opportunities for us to probe the structural and functional importance of APOE variants extensively. <b>Method:</b> In this study, we are taking an ensemble approach that simultaneously uses large-scale protein sequence-based models, including Evolutionary Scale Model and AlphaFold, together with a few <i>in-silico</i> functional prediction web services to investigate the known and possibly disease-causing SAVs in APOE and evaluate their likelihood of being functional and structurally disruptive. <b>Results:</b> As a result, using an ensemble approach with little to no prior field-specific knowledge, we reported 5 SAVs in APOE gene to be potentially disruptive, one of which (C112R) was classificed by previous studies as a key risk factor for AD. <b>Discussion:</b> Our study provided a novel framework to analyze and prioritize the functional and structural importance of SAVs for future experimental and functional validation.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"3 ","pages":"1122559"},"PeriodicalIF":3.9000,"publicationDate":"2023-04-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10117898/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2023.1122559","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
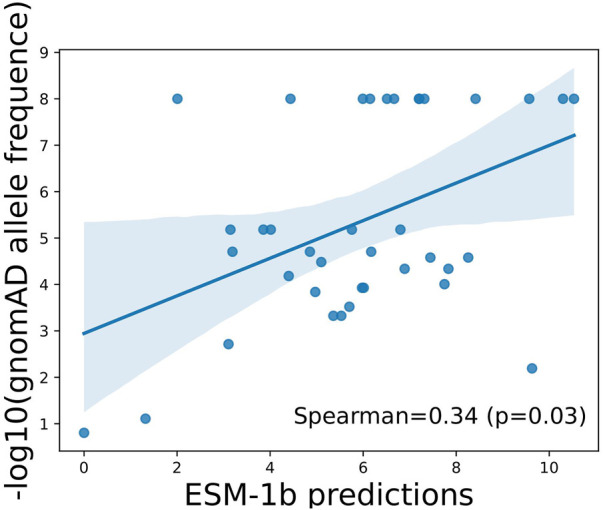
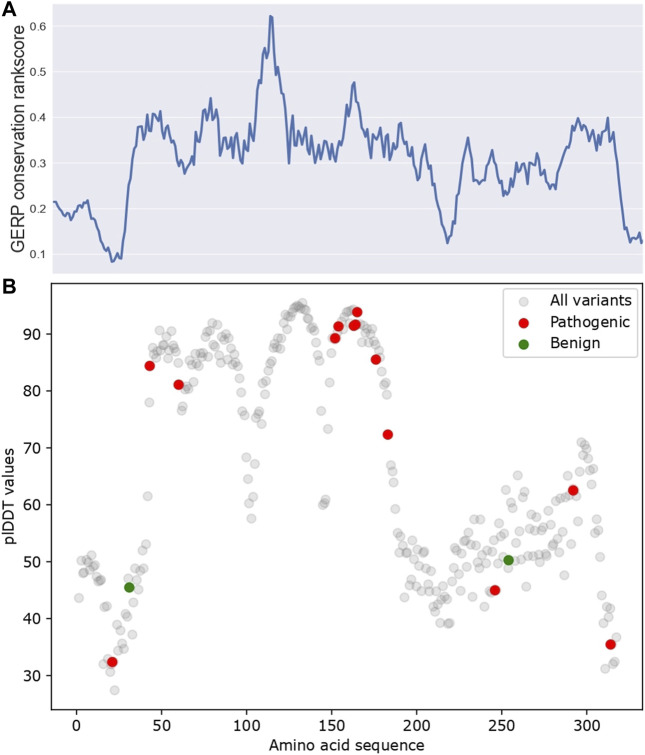
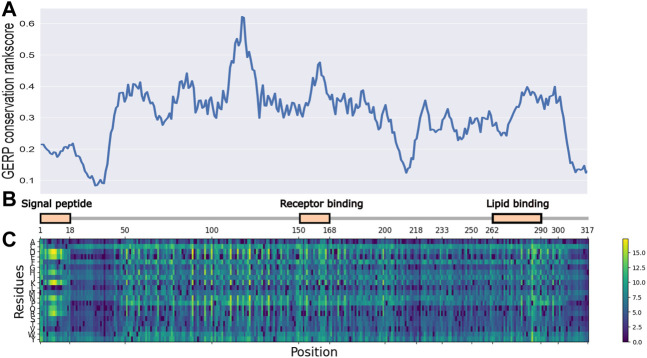
Introduction: Alzheimer's disease (AD) is one of the most prominent medical conditions in the world. Understanding the genetic component of the disease can greatly advance our knowledge regarding its progression, treatment and prognosis. Single amino-acid variants (SAVs) in the APOE gene have been widely investigated as a risk factor for AD Studies, including genome-wide association studies, meta-analysis based studies, and in-vivo animal studies, were carried out to investigate the functional importance and pathogenesis potential of APOE SAVs. However, given the high cost of such large-scale or experimental studies, there are only a handful of variants being reported that have definite explanations. The recent development of in-silico analytical approaches, especially large-scale deep learning models, has opened new opportunities for us to probe the structural and functional importance of APOE variants extensively. Method: In this study, we are taking an ensemble approach that simultaneously uses large-scale protein sequence-based models, including Evolutionary Scale Model and AlphaFold, together with a few in-silico functional prediction web services to investigate the known and possibly disease-causing SAVs in APOE and evaluate their likelihood of being functional and structurally disruptive. Results: As a result, using an ensemble approach with little to no prior field-specific knowledge, we reported 5 SAVs in APOE gene to be potentially disruptive, one of which (C112R) was classificed by previous studies as a key risk factor for AD. Discussion: Our study provided a novel framework to analyze and prioritize the functional and structural importance of SAVs for future experimental and functional validation.
简介阿尔茨海默病(AD)是世界上最常见的疾病之一。了解阿尔茨海默病的遗传因素可极大地促进我们对该病的进展、治疗和预后的了解。APOE 基因中的单氨基酸变异体(SAVs)作为 AD 的风险因素受到了广泛的研究。然而,由于此类大规模研究或实验研究的成本较高,目前报道的变异中只有少数几个能给出明确的解释。近来,体内分析方法的发展,尤其是大规模深度学习模型的发展,为我们广泛探究APOE变异体的结构和功能重要性提供了新的机遇。研究方法在本研究中,我们采用了一种集合方法,同时使用基于大规模蛋白质序列的模型(包括进化尺度模型和 AlphaFold)以及几种内部功能预测网络服务来研究 APOE 中已知的和可能致病的 SAV,并评估它们在功能和结构上具有破坏性的可能性。结果:结果:在几乎没有特定领域知识的情况下,我们使用集合方法报告了 APOE 基因中的 5 个 SAVs 可能具有破坏性,其中一个 SAVs(C112R)被先前的研究归类为 AD 的关键风险因素。讨论我们的研究为今后的实验和功能验证提供了一个新的框架,用于分析 SAVs 的功能和结构重要性并确定其优先次序。


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
