Ji Hye Lee, Ji Sook Yim, Myung Shin Kim, Sin-Young Kim, Shin Hae Park
{"title":"The First Korean Child of Jalili Syndrome with a Novel Missense Mutation in Cation Transport Mediator 4 (CNNM4): A Case Report.","authors":"Ji Hye Lee, Ji Sook Yim, Myung Shin Kim, Sin-Young Kim, Shin Hae Park","doi":"10.3341/kjo.2022.0144","DOIUrl":null,"url":null,"abstract":"Dear Editor, Jalili syndrome is an extremely rare autosomal recessive disorder with two major ocular and dental features: conerod dystrophy and amelogenesis imperfecta [1]. Patients often have poor visual acuity, photophobia, nystagmus, and absent color vision. Since the first case report in 1988, the metal cation transport mediator 4 (CNNM4) gene residing at chromosome locus 2q11.2 is discovered to be a causative gene of Jalili syndrome in 2009 [2,3]. We report the first Korean case of Jalili syndrome carrying compound heterozygous causative variants with a novel missense variant in CNNM4. A 7-year-old boy visited Seoul St. Mary’s Hospital with uncorrected vision. He was the first baby of nonconsanguineous parents and was born at 38 weeks gestation with a birth weight of 3.35 kg. There was no previous family history of ocular, systemic, or chromosomal disorders. He had a mild hyperopic refractive error of +1.5 diopters. He had been wearing glasses for several years, but his best-corrected visual acuity was 20 / 200 in both eyes. Photophobia and slow pendular nystagmus were also observed. Fundus examination revealed a normal-looking optic disc and macula (Fig. 1A, 1B). Whereas widefield fundus photography and fundus autofluorescence did not reveal any definite abnormalities, the blurring of ellipsoid zone was noted on optical coherence tomography (Fig. 1C–1F). A full-field electroretinogram (RETI-scan, Roland Consult) showed severely reduced responses in the cone and rod systems in both eyes (Fig. 1G). After obtaining written informed consent from his parents, next-generation sequencing (NGS) was performed for the genetic analysis. Targeted panel sequencing was done with 439 genes, which were reported as being related to inherited retinal dystrophy. Subsequently, compound heterozygous missense variants of c.1511T>G, p.(Ile504Ser), and c.344T>C, p.(Leu115Pro) in the CNNM4 (reference sequence, NM_020184.4) were identified. The c.1511T>G, p.(Ile504Ser), is in the cystathionine-β-synthase domain which is evolutionarily conserved and known to play essential roles in the regulation of the activities of numerous proteins (PMID: 31347285, 16275737, 14722619). Population frequencies were extremely low (0.0000544 in East Asian populations and 0.00000398 in all ethnicities, according to gnomAD exome), and the variant was predicted to have a deleterious effect by multiple line in silico tools, including MutationTaster [4], PolyPhen-2 [5], and SIFT [6]. Based on these facts, we classified the mutation as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines. A c.344T>C variant has never been reported in a population database. It causes a p.(Leu115Pro) amino acid change, and this position is conserved (phyloP100way, 6.03); multiple in silico tools such as FATHMM-MKL [7], MutationTaster [4], and PolyPhen-2 [5] predicted a deleterious effect by affecting the protein synthetic process. A parental testing, which was performed only for the patient’s mother due to their circumstances, revealed only one heterozygous variant (c.1511T>G) from the mother. Although we could not confirm the other variant from the patient’s father, it is highly suspected that our patient’s variants would be present in trans. On the subsequent dental evaluation, the patient had brown discolorations in his permanent maxillary central incisors and enamel hypoplasia in his permanent first molars. A panoramic radiograph showed pulp obliteration of the remaining primary teeth. Enamel hypoplasia, which is a variant of amelogenesis imperfecta was diagnosed (Fig. 1H). Our case has two major features of Jalili syndrome: cone-rod dystrophy and amelogenesis imperfecta. The mutation in CNNM4 gene was identified by targeted NGS technology. Exact mechanism of CNNM4 mutation has on retina still needs to be uncovered but it seems that as CNNM4 gene encodes an ACDP4 protein which works as a mineral transporter; CNNM4 mutation results in ACDP4 under exKorean J Ophthalmol 2023;37(2):195-197 https://doi.org/10.3341/kjo.2022.0144","PeriodicalId":17883,"journal":{"name":"Korean Journal of Ophthalmology : KJO","volume":"37 2","pages":"195-197"},"PeriodicalIF":0.0000,"publicationDate":"2023-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/54/68/kjo-2022-0144.PMC10151169.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Ophthalmology : KJO","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3341/kjo.2022.0144","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
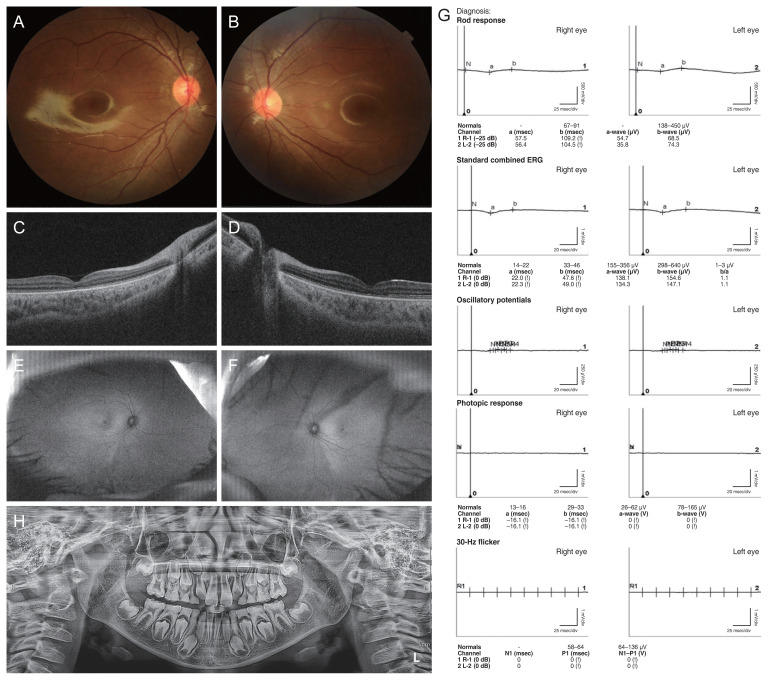
Dear Editor, Jalili syndrome is an extremely rare autosomal recessive disorder with two major ocular and dental features: conerod dystrophy and amelogenesis imperfecta [1]. Patients often have poor visual acuity, photophobia, nystagmus, and absent color vision. Since the first case report in 1988, the metal cation transport mediator 4 (CNNM4) gene residing at chromosome locus 2q11.2 is discovered to be a causative gene of Jalili syndrome in 2009 [2,3]. We report the first Korean case of Jalili syndrome carrying compound heterozygous causative variants with a novel missense variant in CNNM4. A 7-year-old boy visited Seoul St. Mary’s Hospital with uncorrected vision. He was the first baby of nonconsanguineous parents and was born at 38 weeks gestation with a birth weight of 3.35 kg. There was no previous family history of ocular, systemic, or chromosomal disorders. He had a mild hyperopic refractive error of +1.5 diopters. He had been wearing glasses for several years, but his best-corrected visual acuity was 20 / 200 in both eyes. Photophobia and slow pendular nystagmus were also observed. Fundus examination revealed a normal-looking optic disc and macula (Fig. 1A, 1B). Whereas widefield fundus photography and fundus autofluorescence did not reveal any definite abnormalities, the blurring of ellipsoid zone was noted on optical coherence tomography (Fig. 1C–1F). A full-field electroretinogram (RETI-scan, Roland Consult) showed severely reduced responses in the cone and rod systems in both eyes (Fig. 1G). After obtaining written informed consent from his parents, next-generation sequencing (NGS) was performed for the genetic analysis. Targeted panel sequencing was done with 439 genes, which were reported as being related to inherited retinal dystrophy. Subsequently, compound heterozygous missense variants of c.1511T>G, p.(Ile504Ser), and c.344T>C, p.(Leu115Pro) in the CNNM4 (reference sequence, NM_020184.4) were identified. The c.1511T>G, p.(Ile504Ser), is in the cystathionine-β-synthase domain which is evolutionarily conserved and known to play essential roles in the regulation of the activities of numerous proteins (PMID: 31347285, 16275737, 14722619). Population frequencies were extremely low (0.0000544 in East Asian populations and 0.00000398 in all ethnicities, according to gnomAD exome), and the variant was predicted to have a deleterious effect by multiple line in silico tools, including MutationTaster [4], PolyPhen-2 [5], and SIFT [6]. Based on these facts, we classified the mutation as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines. A c.344T>C variant has never been reported in a population database. It causes a p.(Leu115Pro) amino acid change, and this position is conserved (phyloP100way, 6.03); multiple in silico tools such as FATHMM-MKL [7], MutationTaster [4], and PolyPhen-2 [5] predicted a deleterious effect by affecting the protein synthetic process. A parental testing, which was performed only for the patient’s mother due to their circumstances, revealed only one heterozygous variant (c.1511T>G) from the mother. Although we could not confirm the other variant from the patient’s father, it is highly suspected that our patient’s variants would be present in trans. On the subsequent dental evaluation, the patient had brown discolorations in his permanent maxillary central incisors and enamel hypoplasia in his permanent first molars. A panoramic radiograph showed pulp obliteration of the remaining primary teeth. Enamel hypoplasia, which is a variant of amelogenesis imperfecta was diagnosed (Fig. 1H). Our case has two major features of Jalili syndrome: cone-rod dystrophy and amelogenesis imperfecta. The mutation in CNNM4 gene was identified by targeted NGS technology. Exact mechanism of CNNM4 mutation has on retina still needs to be uncovered but it seems that as CNNM4 gene encodes an ACDP4 protein which works as a mineral transporter; CNNM4 mutation results in ACDP4 under exKorean J Ophthalmol 2023;37(2):195-197 https://doi.org/10.3341/kjo.2022.0144



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
