Vitor R C Aguiar, Erick C Castelli, Richard M Single, Arman Bashirova, Veron Ramsuran, Smita Kulkarni, Danillo G Augusto, Maureen P Martin, Maria Gutierrez-Arcelus, Mary Carrington, Diogo Meyer
{"title":"Comparison between qPCR and RNA-seq reveals challenges of quantifying HLA expression.","authors":"Vitor R C Aguiar, Erick C Castelli, Richard M Single, Arman Bashirova, Veron Ramsuran, Smita Kulkarni, Danillo G Augusto, Maureen P Martin, Maria Gutierrez-Arcelus, Mary Carrington, Diogo Meyer","doi":"10.1007/s00251-023-01296-7","DOIUrl":null,"url":null,"abstract":"<p><p>Human leukocyte antigen (HLA) class I and II loci are essential elements of innate and acquired immunity. Their functions include antigen presentation to T cells leading to cellular and humoral immune responses, and modulation of NK cells. Their exceptional influence on disease outcome has now been made clear by genome-wide association studies. The exons encoding the peptide-binding groove have been the main focus for determining HLA effects on disease susceptibility/pathogenesis. However, HLA expression levels have also been implicated in disease outcome, adding another dimension to the extreme diversity of HLA that impacts variability in immune responses across individuals. To estimate HLA expression, immunogenetic studies traditionally rely on quantitative PCR (qPCR). Adoption of alternative high-throughput technologies such as RNA-seq has been hampered by technical issues due to the extreme polymorphism at HLA genes. Recently, however, multiple bioinformatic methods have been developed to accurately estimate HLA expression from RNA-seq data. This opens an exciting opportunity to quantify HLA expression in large datasets but also brings questions on whether RNA-seq results are comparable to those by qPCR. In this study, we analyze three classes of expression data for HLA class I genes for a matched set of individuals: (a) RNA-seq, (b) qPCR, and (c) cell surface HLA-C expression. We observed a moderate correlation between expression estimates from qPCR and RNA-seq for HLA-A, -B, and -C (0.2 ≤ rho ≤ 0.53). We discuss technical and biological factors which need to be accounted for when comparing quantifications for different molecular phenotypes or using different techniques.</p>","PeriodicalId":13446,"journal":{"name":"Immunogenetics","volume":"75 3","pages":"249-262"},"PeriodicalIF":2.9000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9883133/pdf/","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00251-023-01296-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 5
Abstract
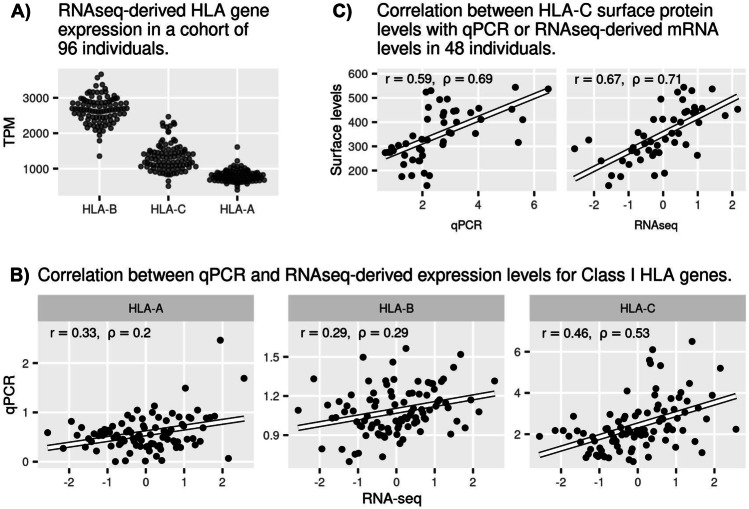
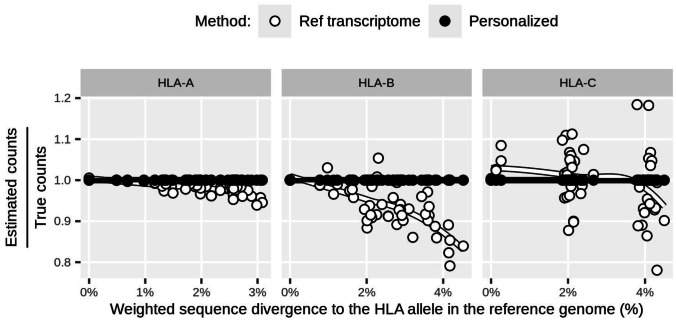
Human leukocyte antigen (HLA) class I and II loci are essential elements of innate and acquired immunity. Their functions include antigen presentation to T cells leading to cellular and humoral immune responses, and modulation of NK cells. Their exceptional influence on disease outcome has now been made clear by genome-wide association studies. The exons encoding the peptide-binding groove have been the main focus for determining HLA effects on disease susceptibility/pathogenesis. However, HLA expression levels have also been implicated in disease outcome, adding another dimension to the extreme diversity of HLA that impacts variability in immune responses across individuals. To estimate HLA expression, immunogenetic studies traditionally rely on quantitative PCR (qPCR). Adoption of alternative high-throughput technologies such as RNA-seq has been hampered by technical issues due to the extreme polymorphism at HLA genes. Recently, however, multiple bioinformatic methods have been developed to accurately estimate HLA expression from RNA-seq data. This opens an exciting opportunity to quantify HLA expression in large datasets but also brings questions on whether RNA-seq results are comparable to those by qPCR. In this study, we analyze three classes of expression data for HLA class I genes for a matched set of individuals: (a) RNA-seq, (b) qPCR, and (c) cell surface HLA-C expression. We observed a moderate correlation between expression estimates from qPCR and RNA-seq for HLA-A, -B, and -C (0.2 ≤ rho ≤ 0.53). We discuss technical and biological factors which need to be accounted for when comparing quantifications for different molecular phenotypes or using different techniques.
期刊介绍:
Immunogenetics publishes original papers, brief communications, and reviews on research in the following areas: genetics and evolution of the immune system; genetic control of immune response and disease susceptibility; bioinformatics of the immune system; structure of immunologically important molecules; and immunogenetics of reproductive biology, tissue differentiation, and development.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
