Marcin P Joachimiak, J Harry Caufield, Nomi L Harris, Hyeongsik Kim, Christopher J Mungall
{"title":"Gene Set Summarization Using Large Language Models.","authors":"Marcin P Joachimiak, J Harry Caufield, Nomi L Harris, Hyeongsik Kim, Christopher J Mungall","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p>Molecular biologists frequently interpret gene lists derived from high-throughput experiments and computational analysis. This is typically done as a statistical enrichment analysis that measures the over- or under-representation of biological function terms associated with genes or their properties, based on curated assertions from a knowledge base (KB) such as the Gene Ontology (GO). Interpreting gene lists can also be framed as a textual summarization task, enabling Large Language Models (LLMs) to use scientific texts directly and avoid reliance on a KB. TALISMAN (Terminological ArtificiaL Intelligence SuMmarization of Annotation and Narratives) uses generative AI to perform gene set function summarization as a complement to standard enrichment analysis. This method can use different sources of gene functional information: (1) structured text derived from curated ontological KB annotations, (2) ontology-free narrative gene summaries, or (3) direct retrieval from the model. We demonstrate that these methods are able to generate plausible and biologically valid summary GO term lists for an input gene set. However, LLM-based approaches are unable to deliver reliable scores or p-values and often return terms that are not statistically significant. Crucially, in our experiments these methods were rarely able to recapitulate the most precise and informative term from standard enrichment analysis. We also observe minor differences depending on prompt input information, with GO term descriptions leading to higher recall but lower precision. However, newer LLM models perform statistically significantly better than the oldest model across all performance metrics, suggesting that future models may lead to further improvements. Overall, the results are nondeterministic, with minor variations in prompt resulting in radically different term lists, true to the stochastic nature of LLMs. Our results show that at this point, LLM-based methods are unsuitable as a replacement for standard term enrichment analysis, however they may provide summarization benefits for implicit knowledge integration across extant but unstandardized knowledge, for large sets of features, and where the amount of information is difficult for humans to process.</p>","PeriodicalId":8425,"journal":{"name":"ArXiv","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-07-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10246080/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ArXiv","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
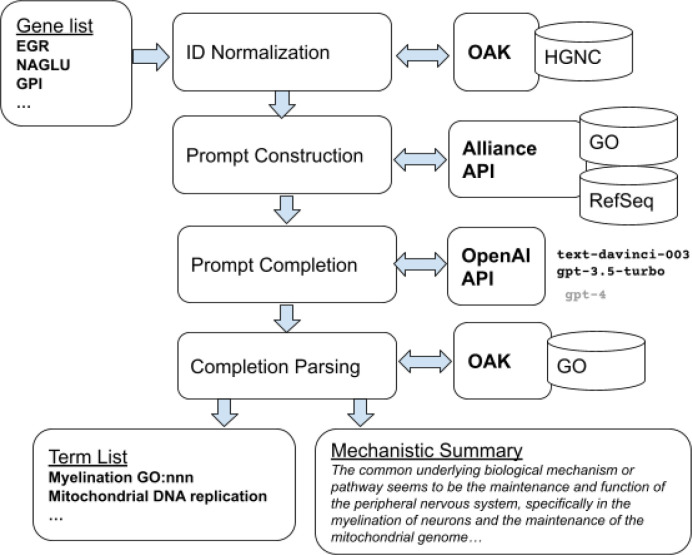
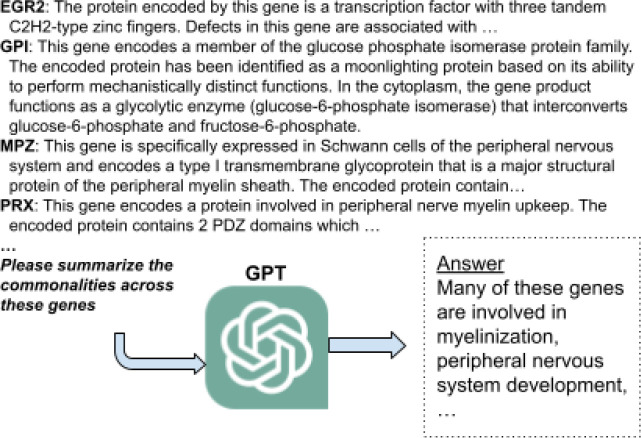
Molecular biologists frequently interpret gene lists derived from high-throughput experiments and computational analysis. This is typically done as a statistical enrichment analysis that measures the over- or under-representation of biological function terms associated with genes or their properties, based on curated assertions from a knowledge base (KB) such as the Gene Ontology (GO). Interpreting gene lists can also be framed as a textual summarization task, enabling Large Language Models (LLMs) to use scientific texts directly and avoid reliance on a KB. TALISMAN (Terminological ArtificiaL Intelligence SuMmarization of Annotation and Narratives) uses generative AI to perform gene set function summarization as a complement to standard enrichment analysis. This method can use different sources of gene functional information: (1) structured text derived from curated ontological KB annotations, (2) ontology-free narrative gene summaries, or (3) direct retrieval from the model. We demonstrate that these methods are able to generate plausible and biologically valid summary GO term lists for an input gene set. However, LLM-based approaches are unable to deliver reliable scores or p-values and often return terms that are not statistically significant. Crucially, in our experiments these methods were rarely able to recapitulate the most precise and informative term from standard enrichment analysis. We also observe minor differences depending on prompt input information, with GO term descriptions leading to higher recall but lower precision. However, newer LLM models perform statistically significantly better than the oldest model across all performance metrics, suggesting that future models may lead to further improvements. Overall, the results are nondeterministic, with minor variations in prompt resulting in radically different term lists, true to the stochastic nature of LLMs. Our results show that at this point, LLM-based methods are unsuitable as a replacement for standard term enrichment analysis, however they may provide summarization benefits for implicit knowledge integration across extant but unstandardized knowledge, for large sets of features, and where the amount of information is difficult for humans to process.

 分享
分享



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
