{"title":"A family with brachydactyly mental retardation syndrome with a missense variant in <i>HDAC4</i>.","authors":"Shinji Takeyari, Kenichi Yamamoto, Makoto Fujiwara, Yasuhisa Ohata, Taichi Kitaoka, Takuo Kubota, Miho Nagata, Yasuki Ishihara, Yohei Miyashita, Yoshihiro Asano, Keiichi Ozono","doi":"10.1297/cpe.2022-0076","DOIUrl":null,"url":null,"abstract":"<p><p>Brachydactyly mental retardation syndrome (BDMR) or chromosome 2q37 deletion syndrome is a genetic disorder caused by 2q37 deletion or haploinsufficiency of histone deacetylase 4 (HDAC4). The <i>HDAC4</i> gene is responsible for major BDMR phenotypes. The symptoms of BDMR include mild-to-moderate intellectual disability, seizures, autism spectrum disorder, short stature, obesity, and facial dysmorphism. Here, we report a family (n = 5) with BDMR who had a missense variant of <i>HDAC4</i>. Four affected individuals [5-yr-old girl (index case); 15- and 3-yr-old siblings; and father] had mild intellectual disability, three of the four affected individuals had short stature and mild cardiac anomalies, and two of the four affected individuals had hypothyroidism. Whole-exome sequencing and analyses of the index case and her family revealed an allelic variant in the <i>HDAC4</i> gene (NM_001378414.1:c.2204G>A:p. Arg735Gln). A healthy family member (mother) did not have the missense variant. To our knowledge, this is the first report of a missense variation in <i>HDAC4</i> that is associated with BDMR.</p>","PeriodicalId":72619,"journal":{"name":"","volume":"32 2","pages":"105-109"},"PeriodicalIF":0.0,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/a7/e8/cpe-32-105.PMC10068624.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2022-0076","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
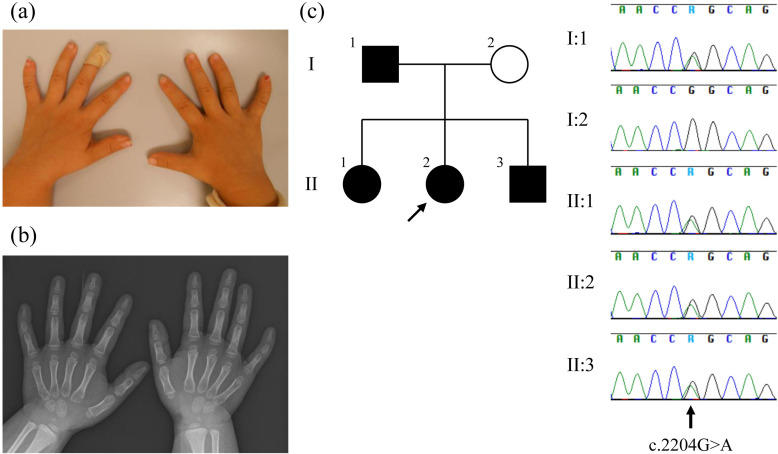
Brachydactyly mental retardation syndrome (BDMR) or chromosome 2q37 deletion syndrome is a genetic disorder caused by 2q37 deletion or haploinsufficiency of histone deacetylase 4 (HDAC4). The HDAC4 gene is responsible for major BDMR phenotypes. The symptoms of BDMR include mild-to-moderate intellectual disability, seizures, autism spectrum disorder, short stature, obesity, and facial dysmorphism. Here, we report a family (n = 5) with BDMR who had a missense variant of HDAC4. Four affected individuals [5-yr-old girl (index case); 15- and 3-yr-old siblings; and father] had mild intellectual disability, three of the four affected individuals had short stature and mild cardiac anomalies, and two of the four affected individuals had hypothyroidism. Whole-exome sequencing and analyses of the index case and her family revealed an allelic variant in the HDAC4 gene (NM_001378414.1:c.2204G>A:p. Arg735Gln). A healthy family member (mother) did not have the missense variant. To our knowledge, this is the first report of a missense variation in HDAC4 that is associated with BDMR.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
