{"title":"Multi-omics Pathways Workflow (MOPAW): An Automated Multi-omics Workflow on the Cancer Genomics Cloud.","authors":"Trinh Nguyen, Xiaopeng Bian, David Roberson, Rakesh Khanna, Qingrong Chen, Chunhua Yan, Rowan Beck, Zelia Worman, Daoud Meerzaman","doi":"10.1177/11769351231180992","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>In the era of big data, gene-set pathway analyses derived from multi-omics are exceptionally powerful. When preparing and analyzing high-dimensional multi-omics data, the installation process and programing skills required to use existing tools can be challenging. This is especially the case for those who are not familiar with coding. In addition, implementation with high performance computing solutions is required to run these tools efficiently.</p><p><strong>Methods: </strong>We introduce an automatic multi-omics pathway workflow, a point and click graphical user interface to Multivariate Single Sample Gene Set Analysis (MOGSA), hosted on the Cancer Genomics Cloud by Seven Bridges Genomics. This workflow leverages the combination of different tools to perform data preparation for each given data types, dimensionality reduction, and MOGSA pathway analysis. The Omics data includes copy number alteration, transcriptomics data, proteomics and phosphoproteomics data. We have also provided an additional workflow to help with downloading data from The Cancer Genome Atlas and Clinical Proteomic Tumor Analysis Consortium and preprocessing these data to be used for this multi-omics pathway workflow.</p><p><strong>Results: </strong>The main outputs of this workflow are the distinct pathways for subgroups of interest provided by users, which are displayed in heatmaps if identified. In addition to this, graphs and tables are provided to users for reviewing.</p><p><strong>Conclusion: </strong>Multi-omics Pathway Workflow requires no coding experience. Users can bring their own data or download and preprocess public datasets from The Cancer Genome Atlas and Clinical Proteomic Tumor Analysis Consortium using our additional workflow based on the samples of interest. Distinct overactivated or deactivated pathways for groups of interest can be found. This useful information is important in effective therapeutic targeting.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"22 ","pages":"11769351231180992"},"PeriodicalIF":2.5000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/28/1c/10.1177_11769351231180992.PMC10278438.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351231180992","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: In the era of big data, gene-set pathway analyses derived from multi-omics are exceptionally powerful. When preparing and analyzing high-dimensional multi-omics data, the installation process and programing skills required to use existing tools can be challenging. This is especially the case for those who are not familiar with coding. In addition, implementation with high performance computing solutions is required to run these tools efficiently.
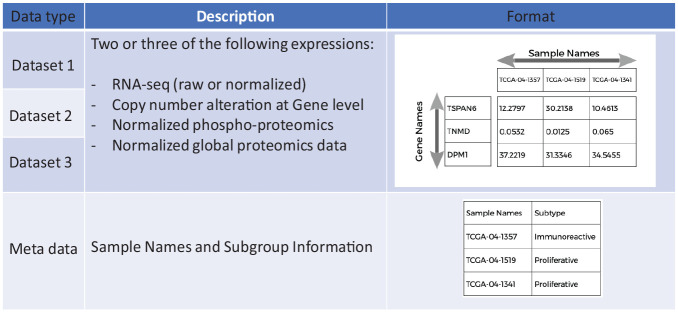
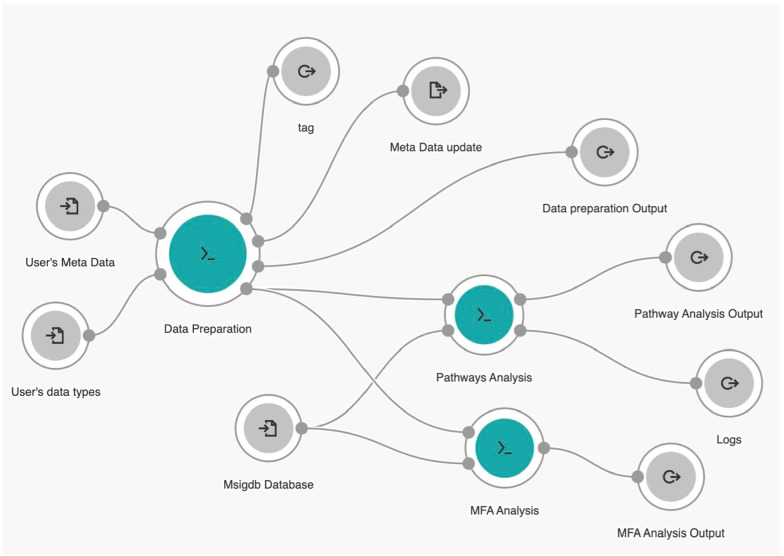
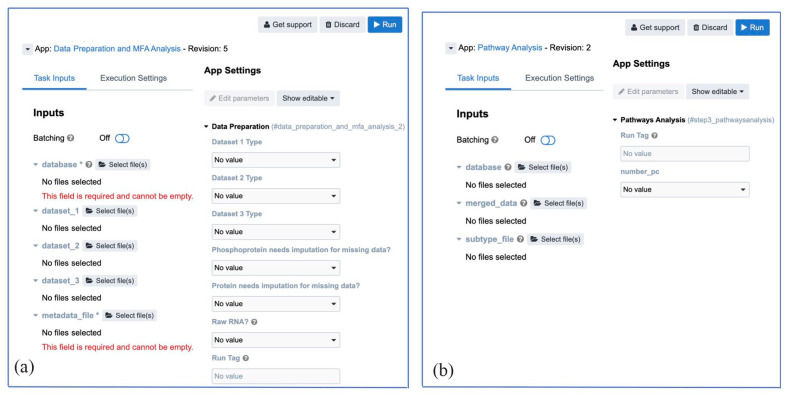
Methods: We introduce an automatic multi-omics pathway workflow, a point and click graphical user interface to Multivariate Single Sample Gene Set Analysis (MOGSA), hosted on the Cancer Genomics Cloud by Seven Bridges Genomics. This workflow leverages the combination of different tools to perform data preparation for each given data types, dimensionality reduction, and MOGSA pathway analysis. The Omics data includes copy number alteration, transcriptomics data, proteomics and phosphoproteomics data. We have also provided an additional workflow to help with downloading data from The Cancer Genome Atlas and Clinical Proteomic Tumor Analysis Consortium and preprocessing these data to be used for this multi-omics pathway workflow.
Results: The main outputs of this workflow are the distinct pathways for subgroups of interest provided by users, which are displayed in heatmaps if identified. In addition to this, graphs and tables are provided to users for reviewing.
Conclusion: Multi-omics Pathway Workflow requires no coding experience. Users can bring their own data or download and preprocess public datasets from The Cancer Genome Atlas and Clinical Proteomic Tumor Analysis Consortium using our additional workflow based on the samples of interest. Distinct overactivated or deactivated pathways for groups of interest can be found. This useful information is important in effective therapeutic targeting.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
