{"title":"Granulomatous Pigmented Purpuric Dermatosis in a Patient with (Inactive) Myasthenia Gravis: A Case Report and Review of the Literature.","authors":"Apirada Dhekariyapak, Penpun Wattanakrai","doi":"10.1159/000530034","DOIUrl":null,"url":null,"abstract":"<p><p>Granulomatous pigmented purpuric dermatosis (GPPD) is a rare histologic variant of pigmented purpuric dermatosis (PPD) characterized by dermal histiocyte-rich interstitial infiltration with or without granuloma formation in addition to the other typical features of PPD. GPPD was previously observed more frequently to affect Asians and was reported to be associated with dyslipidemia. However, our literature search of 45 documented GPPD cases revealed an increasing prevalence in Caucasians in addition to dyslipidemia and associated autoimmune diseases. To date, etiopathogenesis of GPPD is unknown but may involve dyslipidemia, genetic and immunological factors such as autoimmune dysregulation or a sarcoidal reaction associated with <i>C. acnes</i>. GPPD is usually persistent and recalcitrant to treatments. We report a case of GPPD in a 57-year-old Thai woman with underlying myasthenia gravis who presented with a pruritic eruption on both lower legs. After treatment with 0.05% clobetasol propionate cream and oral colchicine, the lesion improved with marked flattening and disappeared with residual postinflammatory hyperpigmentation. We provide a literature review of the epidemiology, etiopathogenesis, concomitant comorbidities, clinical symptoms, dermatoscopic features, and treatments of GPPD.</p>","PeriodicalId":9619,"journal":{"name":"Case Reports in Dermatology","volume":"15 1","pages":"71-84"},"PeriodicalIF":0.8000,"publicationDate":"2023-05-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10293941/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Dermatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000530034","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"DERMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
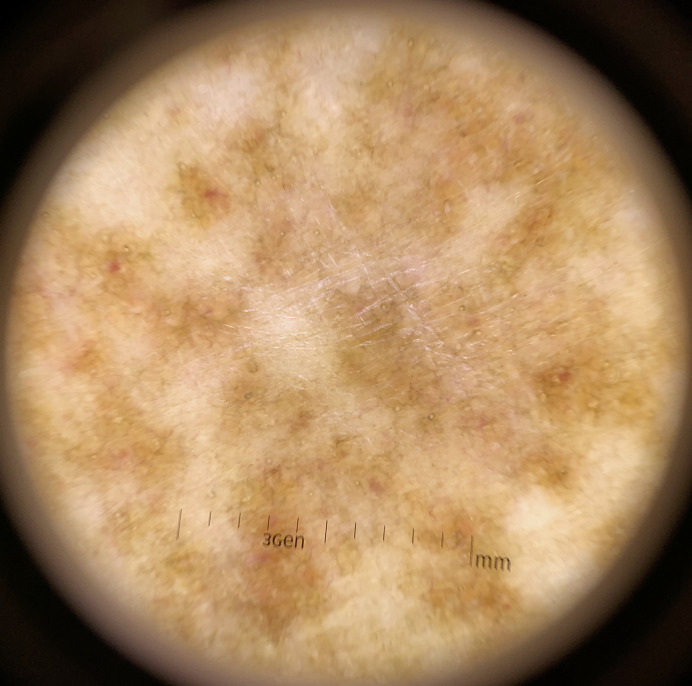
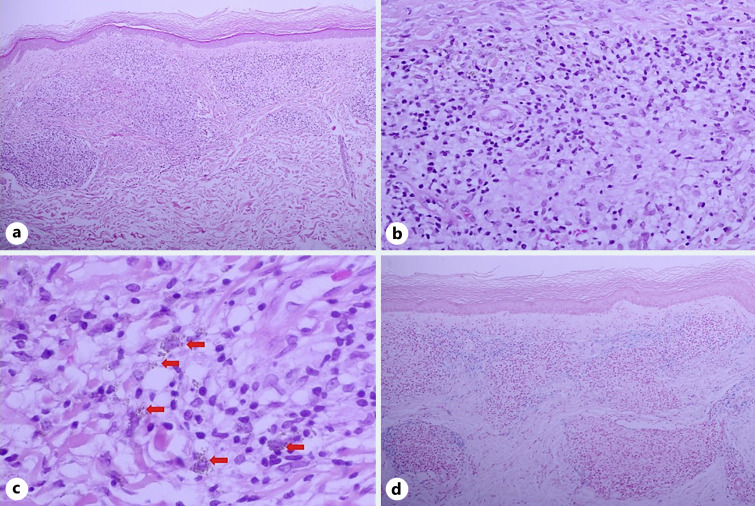
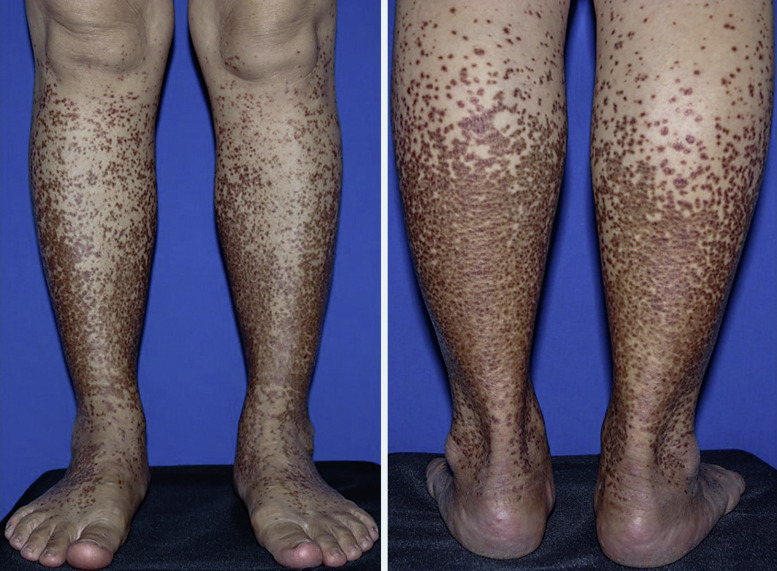
Granulomatous pigmented purpuric dermatosis (GPPD) is a rare histologic variant of pigmented purpuric dermatosis (PPD) characterized by dermal histiocyte-rich interstitial infiltration with or without granuloma formation in addition to the other typical features of PPD. GPPD was previously observed more frequently to affect Asians and was reported to be associated with dyslipidemia. However, our literature search of 45 documented GPPD cases revealed an increasing prevalence in Caucasians in addition to dyslipidemia and associated autoimmune diseases. To date, etiopathogenesis of GPPD is unknown but may involve dyslipidemia, genetic and immunological factors such as autoimmune dysregulation or a sarcoidal reaction associated with C. acnes. GPPD is usually persistent and recalcitrant to treatments. We report a case of GPPD in a 57-year-old Thai woman with underlying myasthenia gravis who presented with a pruritic eruption on both lower legs. After treatment with 0.05% clobetasol propionate cream and oral colchicine, the lesion improved with marked flattening and disappeared with residual postinflammatory hyperpigmentation. We provide a literature review of the epidemiology, etiopathogenesis, concomitant comorbidities, clinical symptoms, dermatoscopic features, and treatments of GPPD.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
