Priscilla S. Seabourn, Danya E. Weber, Helen Spafford, Matthew C. I. Medeiros
{"title":"Aedes albopictus microbiome derives from environmental sources and partitions across distinct host tissues","authors":"Priscilla S. Seabourn, Danya E. Weber, Helen Spafford, Matthew C. I. Medeiros","doi":"10.1002/mbo3.1364","DOIUrl":null,"url":null,"abstract":"<p>The mosquito microbiome consists of a consortium of interacting microorganisms that reside on and within culicid hosts. Mosquitoes acquire most of their microbial diversity from the environment over their life cycle. Once present within the mosquito host, the microbes colonize distinct tissues, and these symbiotic relationships are maintained by immune-related mechanisms, environmental filtering, and trait selection. The processes that govern how environmental microbes assemble across the tissues within mosquitoes remain poorly resolved. We use ecological network analyses to examine how environmental bacteria assemble to form bacteriomes among <i>Aedes albopictus</i> host tissues. Mosquitoes, water, soil, and plant nectar were collected from 20 sites in the Mānoa Valley, Oahu. DNA was extracted and associated bacteriomes were inventoried using Earth Microbiome Project protocols. We find that the bacteriomes of <i>A. albopictus</i> tissues were compositional taxonomic subsets of environmental bacteriomes and suggest that the environmental microbiome serves as a source pool that supports mosquito microbiome diversity. Within the mosquito, the microbiomes of the crop, midgut, Malpighian tubules, and ovaries differed in composition. This microbial diversity partitioned among host tissues formed two specialized modules: one in the crop and midgut, and another in the Malpighian tubules and ovaries. The specialized modules may form based on microbe niche preferences and/or selection of mosquito tissues for specific microbes that aid unique biological functions of the tissue types. A strong niche-driven assembly of tissue-specific microbiotas from the environmental species pool suggests that each tissue has specialized associations with microbes, which derive from host-mediated microbe selection.</p>","PeriodicalId":18573,"journal":{"name":"MicrobiologyOpen","volume":"12 3","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2023-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mbo3.1364","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MicrobiologyOpen","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mbo3.1364","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
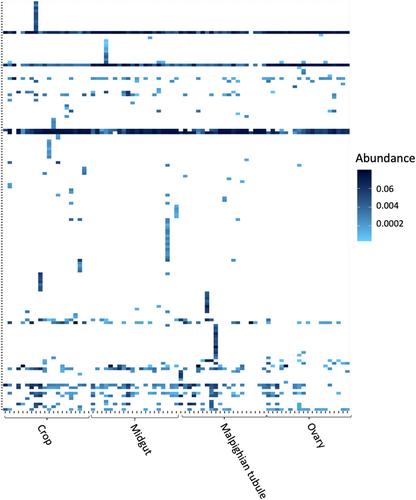
The mosquito microbiome consists of a consortium of interacting microorganisms that reside on and within culicid hosts. Mosquitoes acquire most of their microbial diversity from the environment over their life cycle. Once present within the mosquito host, the microbes colonize distinct tissues, and these symbiotic relationships are maintained by immune-related mechanisms, environmental filtering, and trait selection. The processes that govern how environmental microbes assemble across the tissues within mosquitoes remain poorly resolved. We use ecological network analyses to examine how environmental bacteria assemble to form bacteriomes among Aedes albopictus host tissues. Mosquitoes, water, soil, and plant nectar were collected from 20 sites in the Mānoa Valley, Oahu. DNA was extracted and associated bacteriomes were inventoried using Earth Microbiome Project protocols. We find that the bacteriomes of A. albopictus tissues were compositional taxonomic subsets of environmental bacteriomes and suggest that the environmental microbiome serves as a source pool that supports mosquito microbiome diversity. Within the mosquito, the microbiomes of the crop, midgut, Malpighian tubules, and ovaries differed in composition. This microbial diversity partitioned among host tissues formed two specialized modules: one in the crop and midgut, and another in the Malpighian tubules and ovaries. The specialized modules may form based on microbe niche preferences and/or selection of mosquito tissues for specific microbes that aid unique biological functions of the tissue types. A strong niche-driven assembly of tissue-specific microbiotas from the environmental species pool suggests that each tissue has specialized associations with microbes, which derive from host-mediated microbe selection.
期刊介绍:
MicrobiologyOpen is a peer reviewed, fully open access, broad-scope, and interdisciplinary journal delivering rapid decisions and fast publication of microbial science, a field which is undergoing a profound and exciting evolution in this post-genomic era.
The journal aims to serve the research community by providing a vehicle for authors wishing to publish quality research in both fundamental and applied microbiology. Our goal is to publish articles that stimulate discussion and debate, as well as add to our knowledge base and further the understanding of microbial interactions and microbial processes.
MicrobiologyOpen gives prompt and equal consideration to articles reporting theoretical, experimental, applied, and descriptive work in all aspects of bacteriology, virology, mycology and protistology, including, but not limited to:
- agriculture
- antimicrobial resistance
- astrobiology
- biochemistry
- biotechnology
- cell and molecular biology
- clinical microbiology
- computational, systems, and synthetic microbiology
- environmental science
- evolutionary biology, ecology, and systematics
- food science and technology
- genetics and genomics
- geobiology and earth science
- host-microbe interactions
- infectious diseases
- natural products discovery
- pharmaceutical and medicinal chemistry
- physiology
- plant pathology
- veterinary microbiology
We will consider submissions across unicellular and cell-cluster organisms: prokaryotes (bacteria, archaea) and eukaryotes (fungi, protists, microalgae, lichens), as well as viruses and prions infecting or interacting with microorganisms, plants and animals, including genetic, biochemical, biophysical, bioinformatic and structural analyses.
The journal features Original Articles (including full Research articles, Method articles, and Short Communications), Commentaries, Reviews, and Editorials. Original papers must report well-conducted research with conclusions supported by the data presented in the article. We also support confirmatory research and aim to work with authors to meet reviewer expectations.
MicrobiologyOpen publishes articles submitted directly to the journal and those referred from other Wiley journals.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
