Ian C Smith, Chantal A Pileggi, Ying Wang, Kristin Kernohan, Taila Hartley, Hugh J McMillan, Marcos Loreto Sampaio, Gerd Melkus, John Woulfe, Gaganvir Parmar, Pierre R Bourque, Ari Breiner, Jocelyn Zwicker, C Elizabeth Pringle, Olga Jarinova, Hanns Lochmüller, David A Dyment, Bernard Brais, Kym M Boycott, Siegfried Hekimi, Mary-Ellen Harper, Jodi Warman-Chardon
{"title":"Novel Homozygous Variant in <i>COQ7</i> in Siblings With Hereditary Motor Neuropathy.","authors":"Ian C Smith, Chantal A Pileggi, Ying Wang, Kristin Kernohan, Taila Hartley, Hugh J McMillan, Marcos Loreto Sampaio, Gerd Melkus, John Woulfe, Gaganvir Parmar, Pierre R Bourque, Ari Breiner, Jocelyn Zwicker, C Elizabeth Pringle, Olga Jarinova, Hanns Lochmüller, David A Dyment, Bernard Brais, Kym M Boycott, Siegfried Hekimi, Mary-Ellen Harper, Jodi Warman-Chardon","doi":"10.1212/NXG.0000000000200048","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Coenzyme Q<sub>10</sub> (CoQ<sub>10</sub>) is an important electron carrier and antioxidant. The COQ7 enzyme catalyzes the hydroxylation of 5-demethoxyubiquinone-10 (DMQ<sub>10</sub>), the second-to-last step in the CoQ<sub>10</sub> biosynthesis pathway. We report a consanguineous family presenting with a hereditary motor neuropathy associated with a homozygous c.1A > G p.? variant of <i>COQ7</i> with abnormal CoQ<sub>10</sub> biosynthesis.</p><p><strong>Methods: </strong>Affected family members underwent clinical assessments that included nerve conduction testing, histologic analysis, and MRI. Pathogenicity of the <i>COQ7</i> variant was assessed in cultured fibroblasts and skeletal muscle using a combination of immunoblots, respirometry, and quinone analysis.</p><p><strong>Results: </strong>Three affected siblings, ranging from 12 to 24 years of age, presented with a severe length-dependent motor neuropathy with marked symmetric distal weakness and atrophy with normal sensation. Muscle biopsy of the quadriceps revealed chronic denervation pattern. An MRI examination identified moderate to severe fat infiltration in distal muscles. Exome sequencing demonstrated the homozygous <i>COQ7</i> c.1A > G p.? variant that is expected to bypass the first 38 amino acid residues at the n-terminus, initiating instead with methionine at position 39. This is predicted to cause the loss of the cleavable mitochondrial targeting sequence and 2 additional amino acids, thereby preventing the incorporation and subsequent folding of COQ7 into the inner mitochondrial membrane. Pathogenicity of the <i>COQ7</i> variant was demonstrated by diminished COQ7 and CoQ<sub>10</sub> levels in muscle and fibroblast samples of affected siblings but not in the father, unaffected sibling, or unrelated controls. In addition, fibroblasts from affected siblings had substantial accumulation of DMQ<sub>10</sub>, and maximal mitochondrial respiration was impaired in both fibroblasts and muscle.</p><p><strong>Discussion: </strong>This report describes a new neurologic phenotype of <i>COQ7</i>-related primary CoQ<sub>10</sub> deficiency. Novel aspects of the phenotype presented by this family include pure distal motor neuropathy involvement, as well as the lack of upper motor neuron features, cognitive delay, or sensory involvement in comparison with cases of <i>COQ7</i>-related CoQ<sub>10</sub> deficiency previously reported in the literature.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 1","pages":"e200048"},"PeriodicalIF":3.7000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/65/19/NXG-2022-200051.PMC10108386.pdf","citationCount":"2","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200048","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 2
Abstract
Background and objectives: Coenzyme Q10 (CoQ10) is an important electron carrier and antioxidant. The COQ7 enzyme catalyzes the hydroxylation of 5-demethoxyubiquinone-10 (DMQ10), the second-to-last step in the CoQ10 biosynthesis pathway. We report a consanguineous family presenting with a hereditary motor neuropathy associated with a homozygous c.1A > G p.? variant of COQ7 with abnormal CoQ10 biosynthesis.
Methods: Affected family members underwent clinical assessments that included nerve conduction testing, histologic analysis, and MRI. Pathogenicity of the COQ7 variant was assessed in cultured fibroblasts and skeletal muscle using a combination of immunoblots, respirometry, and quinone analysis.
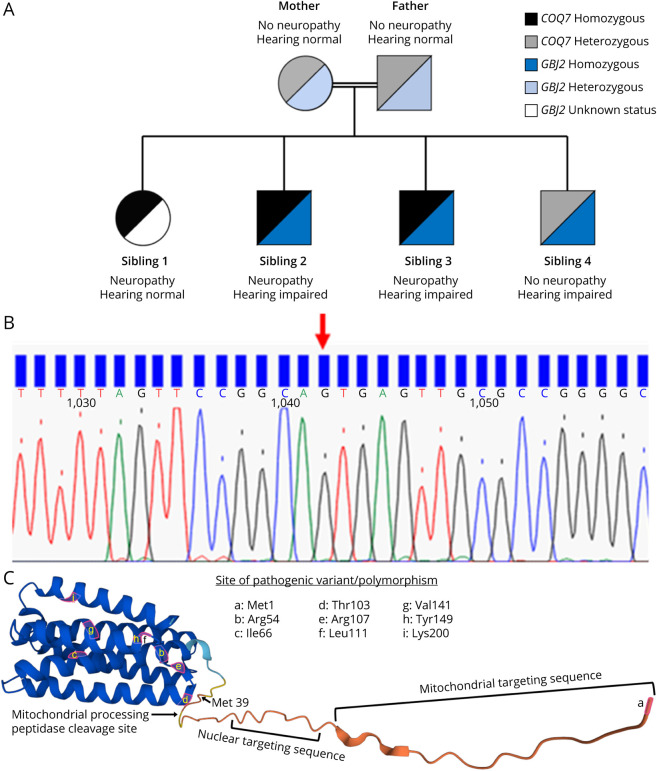
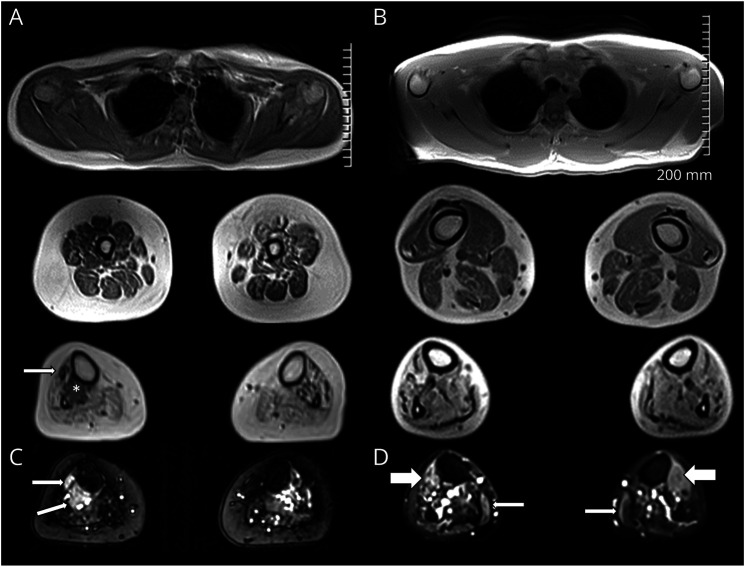
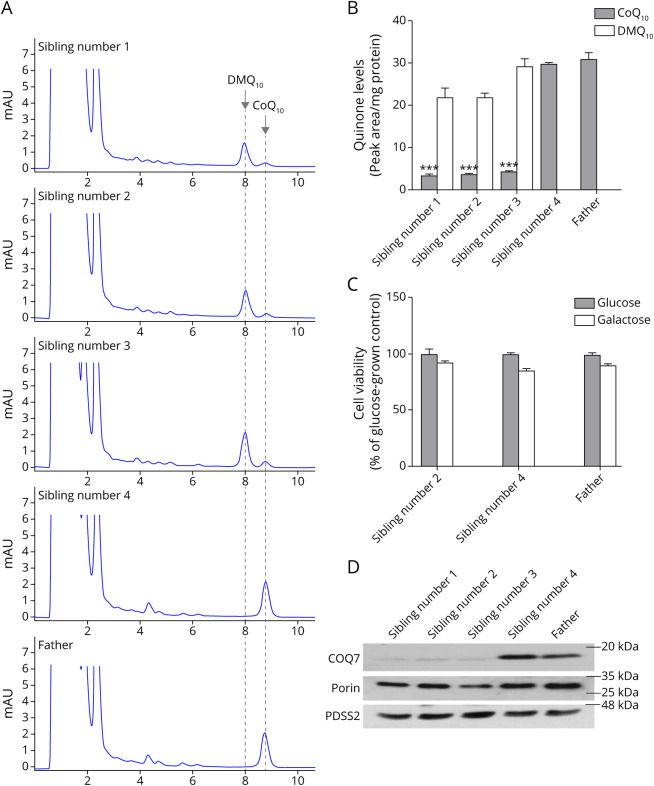
Results: Three affected siblings, ranging from 12 to 24 years of age, presented with a severe length-dependent motor neuropathy with marked symmetric distal weakness and atrophy with normal sensation. Muscle biopsy of the quadriceps revealed chronic denervation pattern. An MRI examination identified moderate to severe fat infiltration in distal muscles. Exome sequencing demonstrated the homozygous COQ7 c.1A > G p.? variant that is expected to bypass the first 38 amino acid residues at the n-terminus, initiating instead with methionine at position 39. This is predicted to cause the loss of the cleavable mitochondrial targeting sequence and 2 additional amino acids, thereby preventing the incorporation and subsequent folding of COQ7 into the inner mitochondrial membrane. Pathogenicity of the COQ7 variant was demonstrated by diminished COQ7 and CoQ10 levels in muscle and fibroblast samples of affected siblings but not in the father, unaffected sibling, or unrelated controls. In addition, fibroblasts from affected siblings had substantial accumulation of DMQ10, and maximal mitochondrial respiration was impaired in both fibroblasts and muscle.
Discussion: This report describes a new neurologic phenotype of COQ7-related primary CoQ10 deficiency. Novel aspects of the phenotype presented by this family include pure distal motor neuropathy involvement, as well as the lack of upper motor neuron features, cognitive delay, or sensory involvement in comparison with cases of COQ7-related CoQ10 deficiency previously reported in the literature.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
