Paloma Gonzalez-Perez, Eleonora S D'Ambrosio, Vincent Picher-Martel, Kathy Chuang, William S David, Anthony A Amato
{"title":"Parent-of-Origin Effect on the Age at Symptom Onset in Myotonic Dystrophy Type 2.","authors":"Paloma Gonzalez-Perez, Eleonora S D'Ambrosio, Vincent Picher-Martel, Kathy Chuang, William S David, Anthony A Amato","doi":"10.1212/NXG.0000000000200073","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>The existence of clinical anticipation, congenital form, and parent-of-origin effect in myotonic dystrophy type 2 (DM2) remains uncertain. Here, we aimed at investigating whether there is a parent-of-origin effect on the age at the first DM2-related clinical manifestation.</p><p><strong>Methods: </strong>We identified patients with genetically confirmed DM2 with known parental inheritance from (1) the electronic medical records of our institutions and (2) a systematic review of the literature following the PRISMA 2020 guidelines and recorded their age at and type of first disease-related symptom. We also interrogated the Myotonic Dystrophy Foundation Family Registry (MDFFR) for patients with DM2 who completed a survey including questions about parental inheritance and age at the first medical problem which they related to their DM2 diagnosis.</p><p><strong>Results: </strong>A total of 26 patients with DM2 from 18 families were identified at our institutions as having maternal (n = 14) or paternal (n = 12) inheritance of the disease, whereas our systematic review of the literature rendered a total of 61 patients with DM2 from 41 families reported by 24 eligible articles as having maternal (n = 40) or paternal (n = 21) inheritance of the disease. Both cohorts were combined for downstream analyses. Up to 61% and 58% of patients had muscle-related symptoms as the first disease manifestation in maternally and paternally inherited DM2 subgroups, respectively. Four patients developed hypotonia at birth and/or delayed motor milestones early in life, and 7 had nonmuscular presentations (2 had cardiac events within the second decade of life and 5 had cataracts), all of them with maternal inheritance. A maternal inheritance was associated with an earlier (within the first 3 decades of life) age at symptom onset relative to a paternal inheritance in this combined cohort, and this association was independent of the patient's sex (OR [95% CI] = 4.245 [1.429-13.820], <i>p</i> = 0.0117). However, this association was not observed in the MDFFR DM2 cohort (n = 127), possibly because age at onset was self-reported, and the information about the type of first symptom or medical problem that patients related to DM2 was lacking.</p><p><strong>Discussion: </strong>A maternal inheritance may increase the risk of an early DM2 onset and of cataracts and cardiovascular events as first DM2 manifestations.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 3","pages":"e200073"},"PeriodicalIF":3.7000,"publicationDate":"2023-04-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/23/f8/NXG-2023-000016.PMC10136683.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200073","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objectives: The existence of clinical anticipation, congenital form, and parent-of-origin effect in myotonic dystrophy type 2 (DM2) remains uncertain. Here, we aimed at investigating whether there is a parent-of-origin effect on the age at the first DM2-related clinical manifestation.
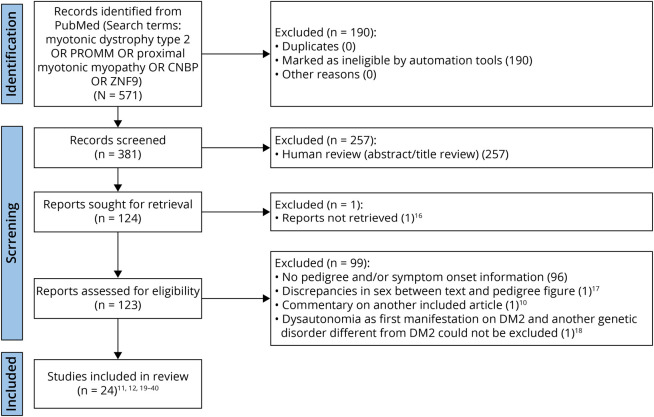
Methods: We identified patients with genetically confirmed DM2 with known parental inheritance from (1) the electronic medical records of our institutions and (2) a systematic review of the literature following the PRISMA 2020 guidelines and recorded their age at and type of first disease-related symptom. We also interrogated the Myotonic Dystrophy Foundation Family Registry (MDFFR) for patients with DM2 who completed a survey including questions about parental inheritance and age at the first medical problem which they related to their DM2 diagnosis.
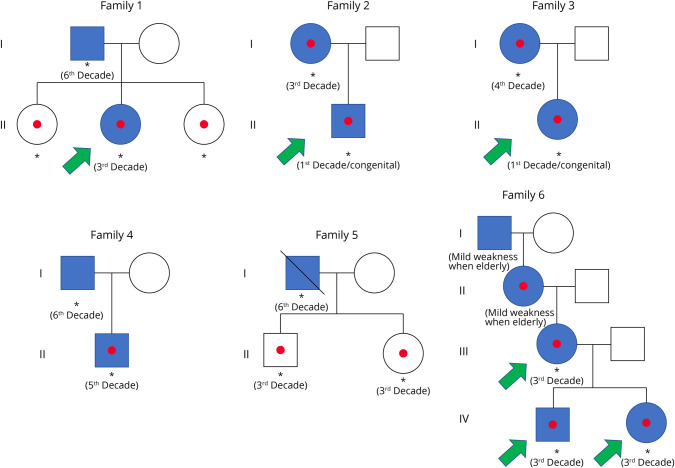
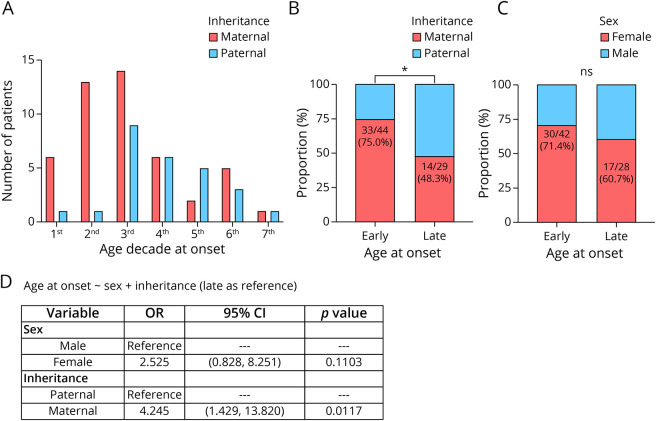
Results: A total of 26 patients with DM2 from 18 families were identified at our institutions as having maternal (n = 14) or paternal (n = 12) inheritance of the disease, whereas our systematic review of the literature rendered a total of 61 patients with DM2 from 41 families reported by 24 eligible articles as having maternal (n = 40) or paternal (n = 21) inheritance of the disease. Both cohorts were combined for downstream analyses. Up to 61% and 58% of patients had muscle-related symptoms as the first disease manifestation in maternally and paternally inherited DM2 subgroups, respectively. Four patients developed hypotonia at birth and/or delayed motor milestones early in life, and 7 had nonmuscular presentations (2 had cardiac events within the second decade of life and 5 had cataracts), all of them with maternal inheritance. A maternal inheritance was associated with an earlier (within the first 3 decades of life) age at symptom onset relative to a paternal inheritance in this combined cohort, and this association was independent of the patient's sex (OR [95% CI] = 4.245 [1.429-13.820], p = 0.0117). However, this association was not observed in the MDFFR DM2 cohort (n = 127), possibly because age at onset was self-reported, and the information about the type of first symptom or medical problem that patients related to DM2 was lacking.
Discussion: A maternal inheritance may increase the risk of an early DM2 onset and of cataracts and cardiovascular events as first DM2 manifestations.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
