{"title":"Is bagging effective in the classification of small-sample genomic and proteomic data?","authors":"T T Vu, U M Braga-Neto","doi":"10.1155/2009/158368","DOIUrl":null,"url":null,"abstract":"<p><p>There has been considerable interest recently in the application of bagging in the classification of both gene-expression data and protein-abundance mass spectrometry data. The approach is often justified by the improvement it produces on the performance of unstable, overfitting classification rules under small-sample situations. However, the question of real practical interest is whether the ensemble scheme will improve performance of those classifiers sufficiently to beat the performance of single stable, nonoverfitting classifiers, in the case of small-sample genomic and proteomic data sets. To investigate that question, we conducted a detailed empirical study, using publicly-available data sets from published genomic and proteomic studies. We observed that, under t-test and RELIEF filter-based feature selection, bagging generally does a good job of improving the performance of unstable, overfitting classifiers, such as CART decision trees and neural networks, but that improvement was not sufficient to beat the performance of single stable, nonoverfitting classifiers, such as diagonal and plain linear discriminant analysis, or 3-nearest neighbors. Furthermore, as expected, the ensemble method did not improve the performance of these classifiers significantly. Representative experimental results are presented and discussed in this work.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":" ","pages":"158368"},"PeriodicalIF":0.0000,"publicationDate":"2009-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2009/158368","citationCount":"10","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2009/158368","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 10
Abstract
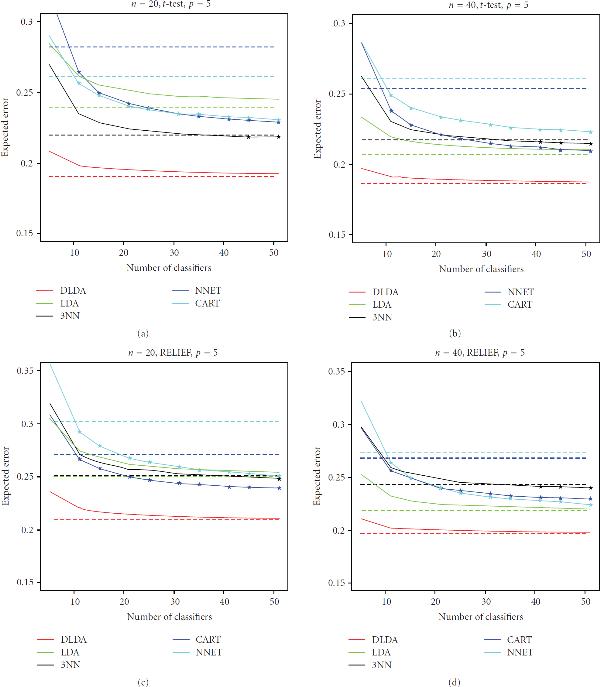
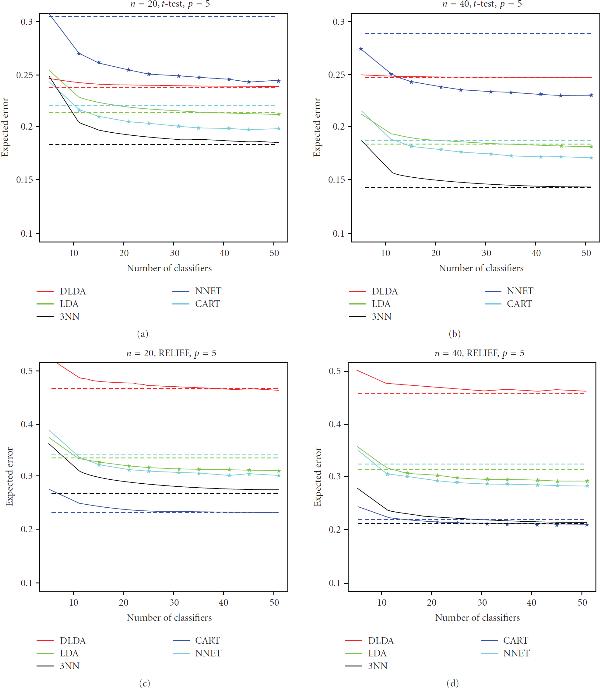
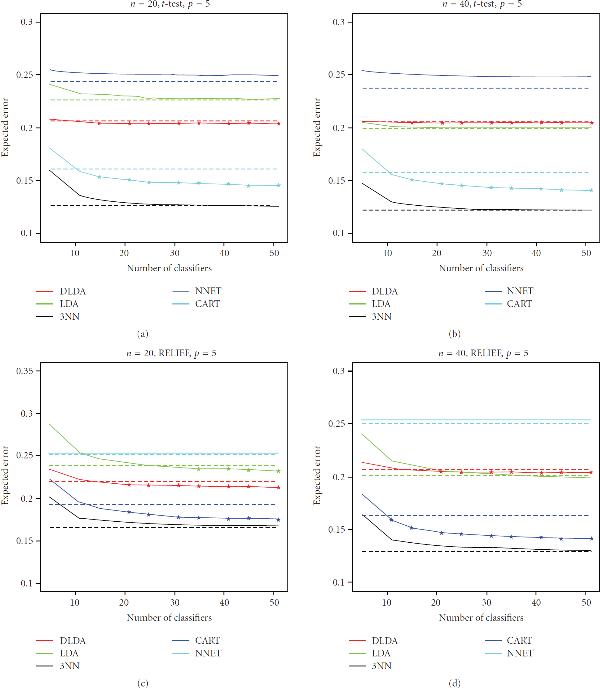
There has been considerable interest recently in the application of bagging in the classification of both gene-expression data and protein-abundance mass spectrometry data. The approach is often justified by the improvement it produces on the performance of unstable, overfitting classification rules under small-sample situations. However, the question of real practical interest is whether the ensemble scheme will improve performance of those classifiers sufficiently to beat the performance of single stable, nonoverfitting classifiers, in the case of small-sample genomic and proteomic data sets. To investigate that question, we conducted a detailed empirical study, using publicly-available data sets from published genomic and proteomic studies. We observed that, under t-test and RELIEF filter-based feature selection, bagging generally does a good job of improving the performance of unstable, overfitting classifiers, such as CART decision trees and neural networks, but that improvement was not sufficient to beat the performance of single stable, nonoverfitting classifiers, such as diagonal and plain linear discriminant analysis, or 3-nearest neighbors. Furthermore, as expected, the ensemble method did not improve the performance of these classifiers significantly. Representative experimental results are presented and discussed in this work.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
