{"title":"Deregulated expression of polycomb repressive complex 2 target genes in a NF1 patient with microdeletion generating the RNF135-SUZ12 chimeric gene.","authors":"Viviana Tritto, Federico Grilli, Donatella Milani, Paola Riva","doi":"10.1007/s10048-023-00718-8","DOIUrl":null,"url":null,"abstract":"<p><p>Neurofibromatosis type I (NF1) microdeletion syndrome, accounting for 5-11% of NF1 patients, is caused by the heterozygous deletion of NF1 and a variable number of flanking genes in the 17q11.2 region. This syndrome is characterized by more severe symptoms than those shown by patients with intragenic NF1 mutation and by variable expressivity, which is not fully explained by the haploinsufficiency of the genes included in the deletions. We here reevaluate an 8-year-old NF1 patient, who carries an atypical deletion generating the RNF135-SUZ12 chimeric gene, previously described when he was 3 years old. As the patient has developed multiple cutaneous/subcutaneous neurofibromas over the past 5 years, we hypothesized a role of RNF135-SUZ12 chimeric gene in the onset of the patient's tumor phenotype. Interestingly, SUZ12 is generally lost or disrupted in NF1 microdeletion syndrome and frequently associated to cancer as RNF135. Expression analysis confirmed the presence of the chimeric gene transcript and revealed hypo-expression of five out of the seven analyzed target genes of the polycomb repressive complex 2 (PRC2), to which SUZ12 belongs, in the patient's peripheral blood, indicating a higher transcriptional repression activity mediated by PRC2. Furthermore, decreased expression of tumor suppressor gene TP53, which is targeted by RNF135, was detected. These results suggest that RNF135-SUZ12 chimera may acquire a gain of function, compared with SUZ12 wild type in the PRC2 complex, and a loss of function relative to RNF135 wild type. Both events may have a role in the early onset of the patient's neurofibromas.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 3","pages":"181-188"},"PeriodicalIF":1.2000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10319651/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00718-8","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/5/5 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
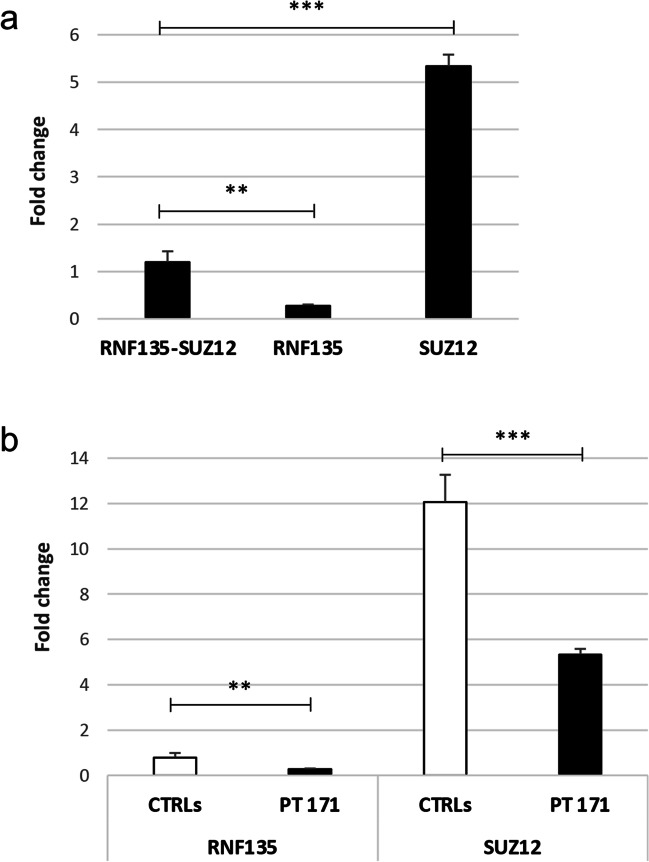
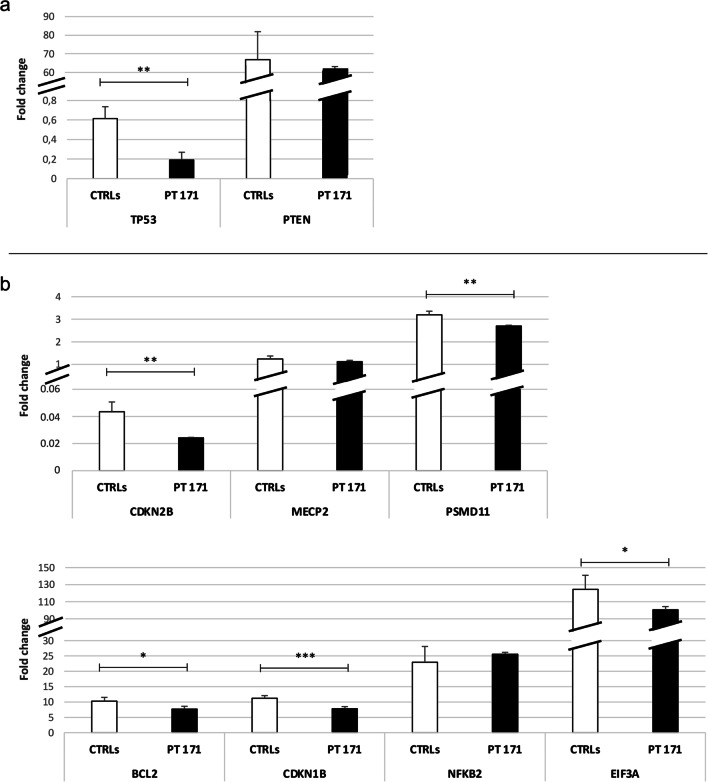
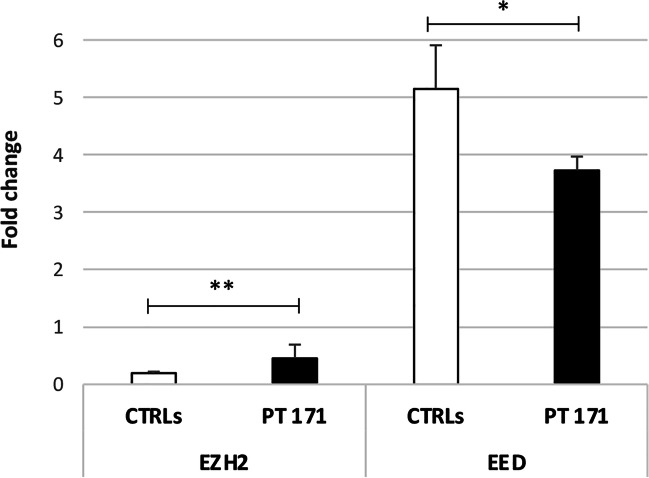
Neurofibromatosis type I (NF1) microdeletion syndrome, accounting for 5-11% of NF1 patients, is caused by the heterozygous deletion of NF1 and a variable number of flanking genes in the 17q11.2 region. This syndrome is characterized by more severe symptoms than those shown by patients with intragenic NF1 mutation and by variable expressivity, which is not fully explained by the haploinsufficiency of the genes included in the deletions. We here reevaluate an 8-year-old NF1 patient, who carries an atypical deletion generating the RNF135-SUZ12 chimeric gene, previously described when he was 3 years old. As the patient has developed multiple cutaneous/subcutaneous neurofibromas over the past 5 years, we hypothesized a role of RNF135-SUZ12 chimeric gene in the onset of the patient's tumor phenotype. Interestingly, SUZ12 is generally lost or disrupted in NF1 microdeletion syndrome and frequently associated to cancer as RNF135. Expression analysis confirmed the presence of the chimeric gene transcript and revealed hypo-expression of five out of the seven analyzed target genes of the polycomb repressive complex 2 (PRC2), to which SUZ12 belongs, in the patient's peripheral blood, indicating a higher transcriptional repression activity mediated by PRC2. Furthermore, decreased expression of tumor suppressor gene TP53, which is targeted by RNF135, was detected. These results suggest that RNF135-SUZ12 chimera may acquire a gain of function, compared with SUZ12 wild type in the PRC2 complex, and a loss of function relative to RNF135 wild type. Both events may have a role in the early onset of the patient's neurofibromas.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
