Tina Eshaghian, Bahareh Rabbani, Reza Shervin Badv, Sahar Mikaeeli, Behdad Gharib, Stanley Iyadurai, Nejat Mahdieh
{"title":"COLQ-related congenital myasthenic syndrome: An integrative view.","authors":"Tina Eshaghian, Bahareh Rabbani, Reza Shervin Badv, Sahar Mikaeeli, Behdad Gharib, Stanley Iyadurai, Nejat Mahdieh","doi":"10.1007/s10048-023-00719-7","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital myasthenic syndromes are inherited disorders caused by mutation in components of the neuromuscular junction and manifest early in life. Mutations in COLQ gene result in congenital myasthenic syndrome. Here, we present the analysis of data from 209 patients from 195 unrelated families highlighting genotype-phenotype correlation. In addition, we describe a COLQ homozygous variant a new patient and discuss it utilizing the Phyre2 and I-TASSER programs. Clinical, molecular genetics, imaging (MRI), and electrodiagnostic (EEG, EMG/NCS) evaluations were performed. Our data showed 89 pathogenic/likely pathogenic variants including 35 missenses, 21 indels, 14 nonsense, 14 splicing, and 5 large deletions variants. Eight common variants were responsible for 48.46% of those. Weakness in proximal muscles, hypotonia, and generalized weakness were detected in all individuals tested. Apart from the weakness, extensive clinical heterogeneity was noted among patients with COLQ-related patients based on their genotypes-those with variants affecting the splice site exhibited more severe clinical features while those with missense variants displayed milder phenotypes, suggesting the role of differential splice variants in multiple functions within the muscle. Analyses and descriptions of these COLQ variants may be helpful in clinical trial readiness and potential development of novel therapies in the setting of established structure-function relationships.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"24 3","pages":"189-200"},"PeriodicalIF":1.2000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-023-00719-7","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/5/25 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
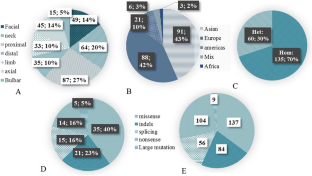
Congenital myasthenic syndromes are inherited disorders caused by mutation in components of the neuromuscular junction and manifest early in life. Mutations in COLQ gene result in congenital myasthenic syndrome. Here, we present the analysis of data from 209 patients from 195 unrelated families highlighting genotype-phenotype correlation. In addition, we describe a COLQ homozygous variant a new patient and discuss it utilizing the Phyre2 and I-TASSER programs. Clinical, molecular genetics, imaging (MRI), and electrodiagnostic (EEG, EMG/NCS) evaluations were performed. Our data showed 89 pathogenic/likely pathogenic variants including 35 missenses, 21 indels, 14 nonsense, 14 splicing, and 5 large deletions variants. Eight common variants were responsible for 48.46% of those. Weakness in proximal muscles, hypotonia, and generalized weakness were detected in all individuals tested. Apart from the weakness, extensive clinical heterogeneity was noted among patients with COLQ-related patients based on their genotypes-those with variants affecting the splice site exhibited more severe clinical features while those with missense variants displayed milder phenotypes, suggesting the role of differential splice variants in multiple functions within the muscle. Analyses and descriptions of these COLQ variants may be helpful in clinical trial readiness and potential development of novel therapies in the setting of established structure-function relationships.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
