Management of Drug Interactions with Inducers: Onset and Disappearance of Induction on Cytochrome P450 3A4 and Uridine Diphosphate Glucuronosyltransferase 1A1 Substrates.
Sara Bettonte, Mattia Berton, Felix Stader, Manuel Battegay, Catia Marzolini
{"title":"Management of Drug Interactions with Inducers: Onset and Disappearance of Induction on Cytochrome P450 3A4 and Uridine Diphosphate Glucuronosyltransferase 1A1 Substrates.","authors":"Sara Bettonte, Mattia Berton, Felix Stader, Manuel Battegay, Catia Marzolini","doi":"10.1007/s13318-023-00833-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>People living with HIV may present co-morbidities requiring the initiation and subsequently the discontinuation of medications with inducing properties. The time to reach maximal enzyme induction and to return to baseline enzyme levels has not been thoroughly characterized.</p><p><strong>Objective: </strong>The aim of this study was to evaluate the onset and disappearance of dolutegravir [uridine diphosphate glucuronosyltransferase (UGT) 1A1 and cytochrome P450 (CYP) 3A4 substrate] and raltegravir (UGT1A1 substrate) induction with strong and moderate inducers using physiologically based pharmacokinetic (PBPK) modeling.</p><p><strong>Methods: </strong>The predictive performance of the PBPK model to simulate dolutegravir and raltegravir pharmacokinetics and to reproduce the strength of induction was verified using clinical drug-drug interaction studies (steady-state induction) and switch studies (residual induction). The model was considered verified when the predictions were within 2-fold of the observed data. One hundred virtual individuals (50% female) were generated to simulate the unstudied scenarios. The results were used to calculate the fold-change in CYP3A4 and UGT1A1 enzyme levels upon initiation and discontinuation of strong (rifampicin) or moderate (efavirenz or rifabutin) inducers.</p><p><strong>Results: </strong>The time for reaching maximal induction and subsequent disappearance of CYP3A4 induction was 14 days for rifampicin and efavirenz but 7 days for rifabutin. The distinct timelines for the moderate inducers relate to their different half-lives and plasma concentrations. The induction and de-induction processes were more rapid for UGT1A1.</p><p><strong>Conclusions: </strong>Our simulations support the common practice of maintaining the adjusted dosage of a drug for another 2 weeks after stopping an inducer. Furthermore, our simulations suggest that an inducer should be administered for at least 14 days before conducting interaction studies to reach maximal induction.</p>","PeriodicalId":11939,"journal":{"name":"European Journal of Drug Metabolism and Pharmacokinetics","volume":"48 4","pages":"353-362"},"PeriodicalIF":2.4000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/6c/b0/13318_2023_Article_833.PMC10322778.pdf","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Drug Metabolism and Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s13318-023-00833-9","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 1
Abstract
Background: People living with HIV may present co-morbidities requiring the initiation and subsequently the discontinuation of medications with inducing properties. The time to reach maximal enzyme induction and to return to baseline enzyme levels has not been thoroughly characterized.
Objective: The aim of this study was to evaluate the onset and disappearance of dolutegravir [uridine diphosphate glucuronosyltransferase (UGT) 1A1 and cytochrome P450 (CYP) 3A4 substrate] and raltegravir (UGT1A1 substrate) induction with strong and moderate inducers using physiologically based pharmacokinetic (PBPK) modeling.
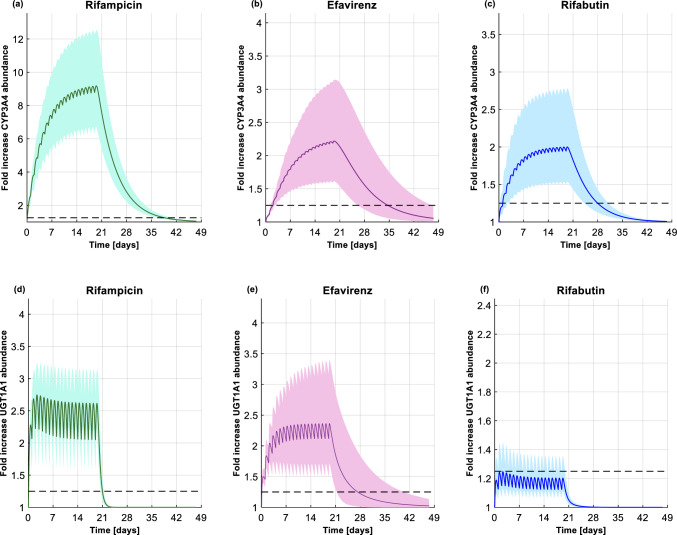
Methods: The predictive performance of the PBPK model to simulate dolutegravir and raltegravir pharmacokinetics and to reproduce the strength of induction was verified using clinical drug-drug interaction studies (steady-state induction) and switch studies (residual induction). The model was considered verified when the predictions were within 2-fold of the observed data. One hundred virtual individuals (50% female) were generated to simulate the unstudied scenarios. The results were used to calculate the fold-change in CYP3A4 and UGT1A1 enzyme levels upon initiation and discontinuation of strong (rifampicin) or moderate (efavirenz or rifabutin) inducers.
Results: The time for reaching maximal induction and subsequent disappearance of CYP3A4 induction was 14 days for rifampicin and efavirenz but 7 days for rifabutin. The distinct timelines for the moderate inducers relate to their different half-lives and plasma concentrations. The induction and de-induction processes were more rapid for UGT1A1.
Conclusions: Our simulations support the common practice of maintaining the adjusted dosage of a drug for another 2 weeks after stopping an inducer. Furthermore, our simulations suggest that an inducer should be administered for at least 14 days before conducting interaction studies to reach maximal induction.
期刊介绍:
Hepatology International is a peer-reviewed journal featuring articles written by clinicians, clinical researchers and basic scientists is dedicated to research and patient care issues in hepatology. This journal focuses mainly on new and emerging diagnostic and treatment options, protocols and molecular and cellular basis of disease pathogenesis, new technologies, in liver and biliary sciences.
Hepatology International publishes original research articles related to clinical care and basic research; review articles; consensus guidelines for diagnosis and treatment; invited editorials, and controversies in contemporary issues. The journal does not publish case reports.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
