Kritika M Garg, Vinita Lamba, Balaji Chattopadhyay
{"title":"Genomic Insights Into the Evolution and Demographic History of the SARS-CoV-2 Omicron Variant: Population Genomics Approach.","authors":"Kritika M Garg, Vinita Lamba, Balaji Chattopadhyay","doi":"10.2196/40673","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>A thorough understanding of the patterns of genetic subdivision in a pathogen can provide crucial information that is necessary to prevent disease spread. For SARS-CoV-2, the availability of millions of genomes makes this task analytically challenging, and traditional methods for understanding genetic subdivision often fail.</p><p><strong>Objective: </strong>The aim of our study was to use population genomics methods to identify the subtle subdivisions and demographic history of the Omicron variant, in addition to those captured by the Pango lineage.</p><p><strong>Methods: </strong>We used a combination of an evolutionary network approach and multivariate statistical protocols to understand the subdivision and spread of the Omicron variant. We identified subdivisions within the BA.1 and BA.2 lineages and further identified the mutations associated with each cluster. We further characterized the overall genomic diversity of the Omicron variant and assessed the selection pressure for each of the genetic clusters identified.</p><p><strong>Results: </strong>We observed concordant results, using two different methods to understand genetic subdivision. The overall pattern of subdivision in the Omicron variant was in broad agreement with the Pango lineage definition. Further, 1 cluster of the BA.1 lineage and 3 clusters of the BA.2 lineage revealed statistically significant signatures of selection or demographic expansion (Tajima's D<-2), suggesting the role of microevolutionary processes in the spread of the virus.</p><p><strong>Conclusions: </strong>We provide an easy framework for assessing the genetic structure and demographic history of SARS-CoV-2, which can be particularly useful for understanding the local history of the virus. We identified important mutations that are advantageous to some lineages of Omicron and aid in the transmission of the virus. This is crucial information for policy makers, as preventive measures can be designed to mitigate further spread based on a holistic understanding of the variability of the virus and the evolutionary processes aiding its spread.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":"4 ","pages":"e40673"},"PeriodicalIF":0.0000,"publicationDate":"2023-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10331448/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/40673","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: A thorough understanding of the patterns of genetic subdivision in a pathogen can provide crucial information that is necessary to prevent disease spread. For SARS-CoV-2, the availability of millions of genomes makes this task analytically challenging, and traditional methods for understanding genetic subdivision often fail.
Objective: The aim of our study was to use population genomics methods to identify the subtle subdivisions and demographic history of the Omicron variant, in addition to those captured by the Pango lineage.
Methods: We used a combination of an evolutionary network approach and multivariate statistical protocols to understand the subdivision and spread of the Omicron variant. We identified subdivisions within the BA.1 and BA.2 lineages and further identified the mutations associated with each cluster. We further characterized the overall genomic diversity of the Omicron variant and assessed the selection pressure for each of the genetic clusters identified.
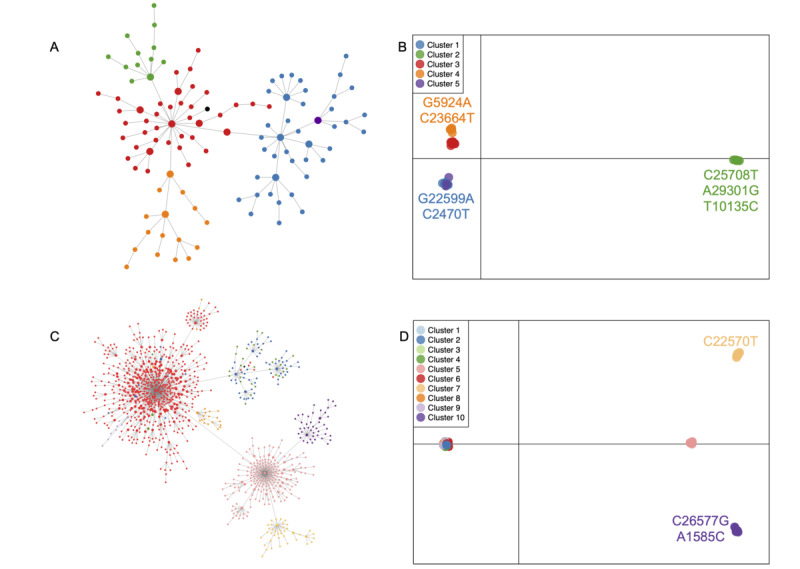
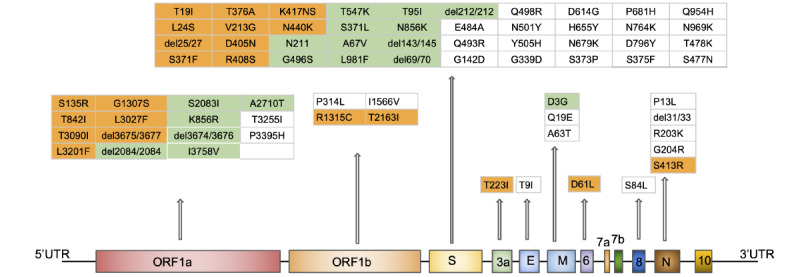
Results: We observed concordant results, using two different methods to understand genetic subdivision. The overall pattern of subdivision in the Omicron variant was in broad agreement with the Pango lineage definition. Further, 1 cluster of the BA.1 lineage and 3 clusters of the BA.2 lineage revealed statistically significant signatures of selection or demographic expansion (Tajima's D<-2), suggesting the role of microevolutionary processes in the spread of the virus.
Conclusions: We provide an easy framework for assessing the genetic structure and demographic history of SARS-CoV-2, which can be particularly useful for understanding the local history of the virus. We identified important mutations that are advantageous to some lineages of Omicron and aid in the transmission of the virus. This is crucial information for policy makers, as preventive measures can be designed to mitigate further spread based on a holistic understanding of the variability of the virus and the evolutionary processes aiding its spread.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
