Adwoa P Boakye-Yiadom, Samuel B Nguah, Haruna Mahama, Gyikua Plange-Rhule
{"title":"Congenital complete arhinia with alobar holoprosencephaly.","authors":"Adwoa P Boakye-Yiadom, Samuel B Nguah, Haruna Mahama, Gyikua Plange-Rhule","doi":"10.4314/gmj.v56i3.14","DOIUrl":null,"url":null,"abstract":"<p><p>Congenital arhinia is a life-threatening, rare craniofacial disorder, which, when not identified and managed early can cause severe respiratory distress at birth due to upper airway obstruction. Since neonates are obligate nasal breathers, simultaneous sucking and breathing requirement in neonates with arhinia leads to respiratory distress. Inspiration and expiration through the oral passage alone may result in thoracic retraction, thereby further exacerbating respiratory distress. We report a rare case of congenital complete arhinia with alobar holoprosencephaly in a 9-month-old. With no family history of congenital malformations, maternal risk factors and uneventful pregnancy, a term female neonate was delivered vaginally without immediate post-delivery respiratory distress. Examination revealed microcephaly, absent fontanelles, fused cranial sutures and bilateral microphthalmia. Breathing was spontaneous, with no immediate signs of respiratory distress. An additional diagnosis of alobar holoprosencephaly was made after a head computed tomography (CT) scan was done. Management included the initial stabilisation phase of supplemental oxygen and an orogastric tube for feeding. The baby did not require both tracheostomy and gastrostomy tubes, as she was not in severe respiratory distress requiring a tracheostomy tube nor having difficulties feeding with the orogastric tube.</p>","PeriodicalId":35509,"journal":{"name":"Ghana Medical Journal","volume":"56 3","pages":"231-235"},"PeriodicalIF":0.0000,"publicationDate":"2022-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10336630/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Ghana Medical Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4314/gmj.v56i3.14","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
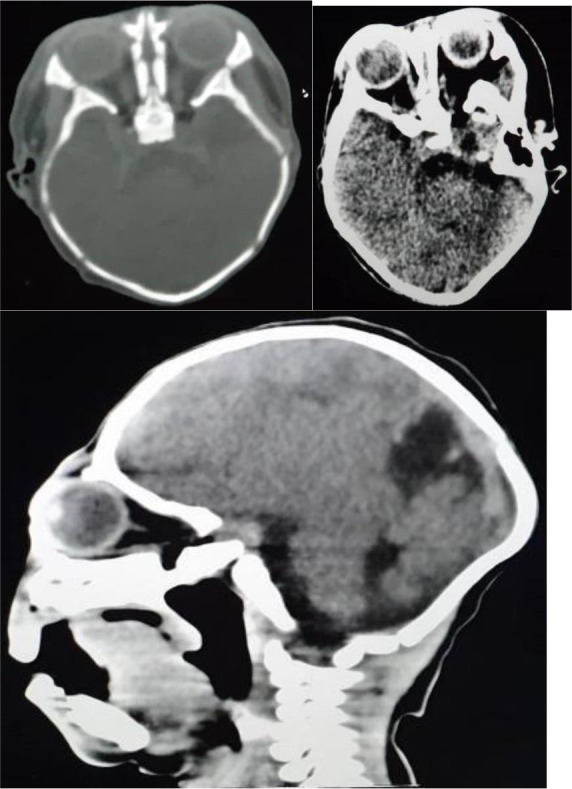
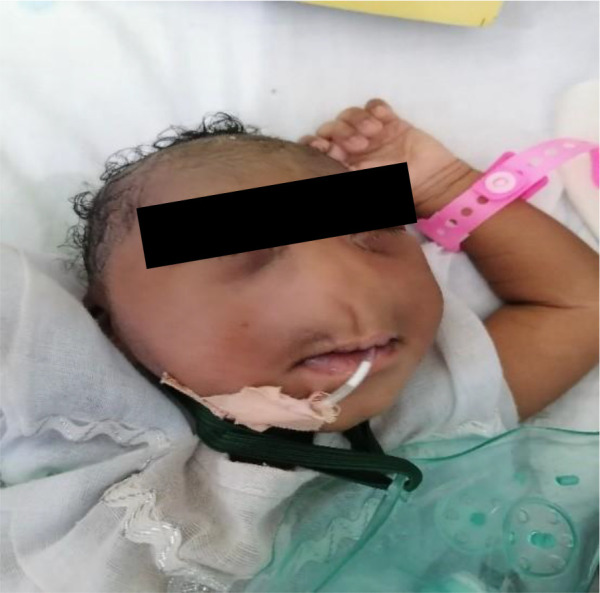
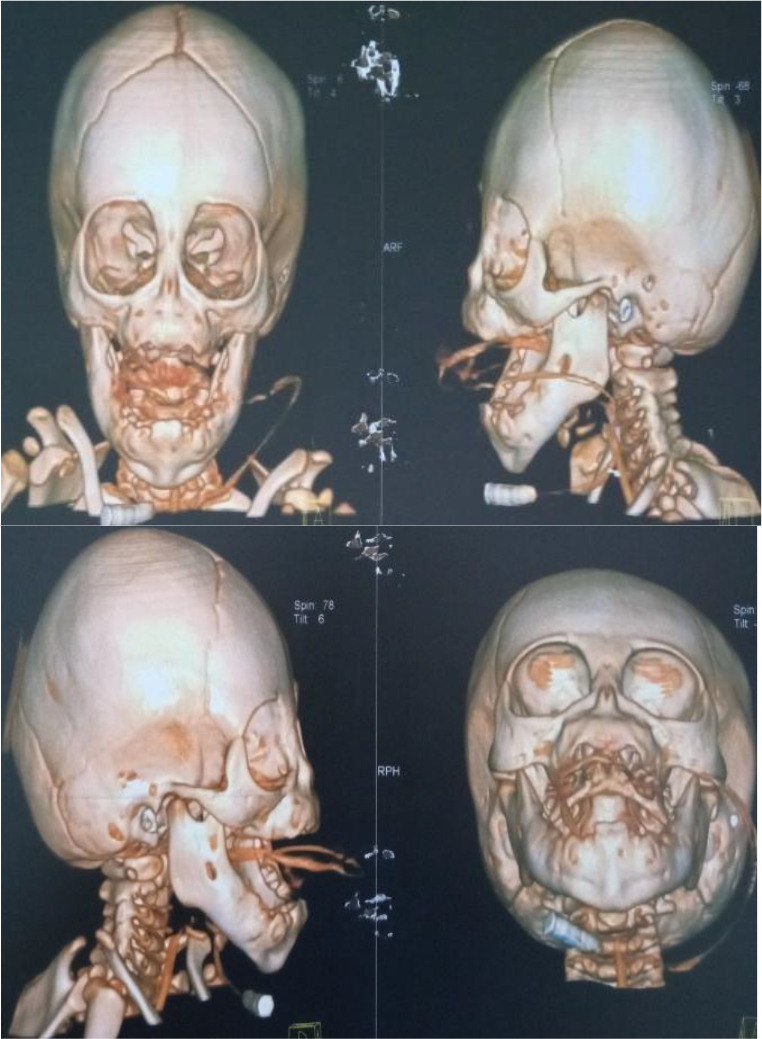
Congenital arhinia is a life-threatening, rare craniofacial disorder, which, when not identified and managed early can cause severe respiratory distress at birth due to upper airway obstruction. Since neonates are obligate nasal breathers, simultaneous sucking and breathing requirement in neonates with arhinia leads to respiratory distress. Inspiration and expiration through the oral passage alone may result in thoracic retraction, thereby further exacerbating respiratory distress. We report a rare case of congenital complete arhinia with alobar holoprosencephaly in a 9-month-old. With no family history of congenital malformations, maternal risk factors and uneventful pregnancy, a term female neonate was delivered vaginally without immediate post-delivery respiratory distress. Examination revealed microcephaly, absent fontanelles, fused cranial sutures and bilateral microphthalmia. Breathing was spontaneous, with no immediate signs of respiratory distress. An additional diagnosis of alobar holoprosencephaly was made after a head computed tomography (CT) scan was done. Management included the initial stabilisation phase of supplemental oxygen and an orogastric tube for feeding. The baby did not require both tracheostomy and gastrostomy tubes, as she was not in severe respiratory distress requiring a tracheostomy tube nor having difficulties feeding with the orogastric tube.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
