An integrated complete-genome sequencing and systems biology approach to predict antimicrobial resistance genes in the virulent bacterial strains of Moraxella catarrhalis.
Sadia Afrin Bristy, Md Arju Hossain, Md Imran Hasan, S M Hasan Mahmud, Mohammad Ali Moni, Md Habibur Rahman
{"title":"An integrated complete-genome sequencing and systems biology approach to predict antimicrobial resistance genes in the virulent bacterial strains of Moraxella catarrhalis.","authors":"Sadia Afrin Bristy, Md Arju Hossain, Md Imran Hasan, S M Hasan Mahmud, Mohammad Ali Moni, Md Habibur Rahman","doi":"10.1093/bfgp/elad005","DOIUrl":null,"url":null,"abstract":"<p><p>Moraxella catarrhalis is a symbiotic as well as mucosal infection-causing bacterium unique to humans. Currently, it is considered as one of the leading factors of acute middle ear infection in children. As M. catarrhalis is resistant to multiple drugs, the treatment is unsuccessful; therefore, innovative and forward-thinking approaches are required to combat the problem of antimicrobial resistance (AMR). To better comprehend the numerous processes that lead to antibiotic resistance in M. catarrhalis, we have adopted a computational method in this study. From the NCBI-Genome database, we investigated 12 strains of M. catarrhalis. We explored the interaction network comprising 74 antimicrobial-resistant genes found by analyzing M. catarrhalis bacterial strains. Moreover, to elucidate the molecular mechanism of the AMR system, clustering and the functional enrichment analysis were assessed employing AMR gene interactions networks. According to the findings of our assessment, the majority of the genes in the network were involved in antibiotic inactivation; antibiotic target replacement, alteration and antibiotic efflux pump processes. They exhibit resistance to several antibiotics, such as isoniazid, ethionamide, cycloserine, fosfomycin, triclosan, etc. Additionally, rpoB, atpA, fusA, groEL and rpoL have the highest frequency of relevant interactors in the interaction network and are therefore regarded as the hub nodes. These genes can be exploited to create novel medications by serving as possible therapeutic targets. Finally, we believe that our findings could be useful to advance knowledge of the AMR system present in M. catarrhalis.</p>","PeriodicalId":55323,"journal":{"name":"Briefings in Functional Genomics","volume":"22 4","pages":"375-391"},"PeriodicalIF":2.5000,"publicationDate":"2023-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10350830/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in Functional Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bfgp/elad005","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
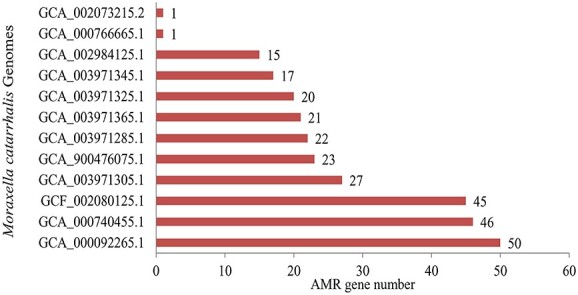
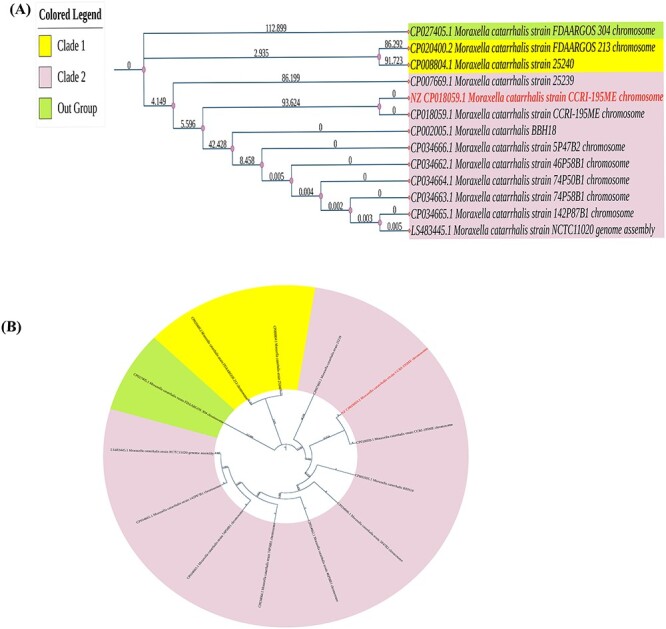
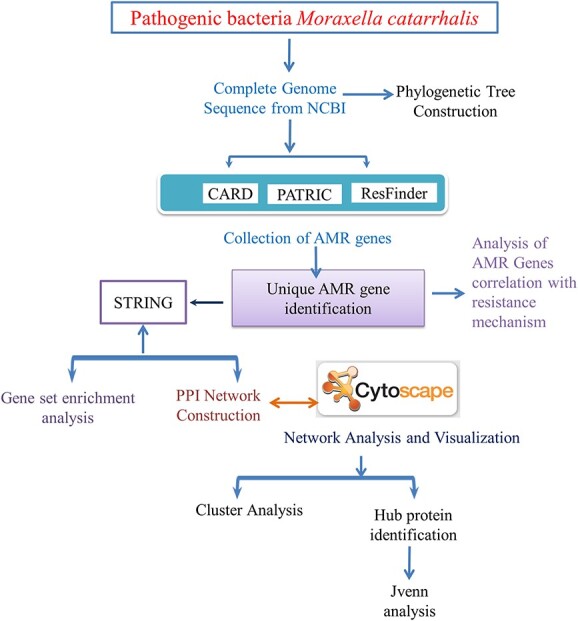
Moraxella catarrhalis is a symbiotic as well as mucosal infection-causing bacterium unique to humans. Currently, it is considered as one of the leading factors of acute middle ear infection in children. As M. catarrhalis is resistant to multiple drugs, the treatment is unsuccessful; therefore, innovative and forward-thinking approaches are required to combat the problem of antimicrobial resistance (AMR). To better comprehend the numerous processes that lead to antibiotic resistance in M. catarrhalis, we have adopted a computational method in this study. From the NCBI-Genome database, we investigated 12 strains of M. catarrhalis. We explored the interaction network comprising 74 antimicrobial-resistant genes found by analyzing M. catarrhalis bacterial strains. Moreover, to elucidate the molecular mechanism of the AMR system, clustering and the functional enrichment analysis were assessed employing AMR gene interactions networks. According to the findings of our assessment, the majority of the genes in the network were involved in antibiotic inactivation; antibiotic target replacement, alteration and antibiotic efflux pump processes. They exhibit resistance to several antibiotics, such as isoniazid, ethionamide, cycloserine, fosfomycin, triclosan, etc. Additionally, rpoB, atpA, fusA, groEL and rpoL have the highest frequency of relevant interactors in the interaction network and are therefore regarded as the hub nodes. These genes can be exploited to create novel medications by serving as possible therapeutic targets. Finally, we believe that our findings could be useful to advance knowledge of the AMR system present in M. catarrhalis.
期刊介绍:
Briefings in Functional Genomics publishes high quality peer reviewed articles that focus on the use, development or exploitation of genomic approaches, and their application to all areas of biological research. As well as exploring thematic areas where these techniques and protocols are being used, articles review the impact that these approaches have had, or are likely to have, on their field. Subjects covered by the Journal include but are not restricted to: the identification and functional characterisation of coding and non-coding features in genomes, microarray technologies, gene expression profiling, next generation sequencing, pharmacogenomics, phenomics, SNP technologies, transgenic systems, mutation screens and genotyping. Articles range in scope and depth from the introductory level to specific details of protocols and analyses, encompassing bacterial, fungal, plant, animal and human data.
The editorial board welcome the submission of review articles for publication. Essential criteria for the publication of papers is that they do not contain primary data, and that they are high quality, clearly written review articles which provide a balanced, highly informative and up to date perspective to researchers in the field of functional genomics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
