Gorka Fernández-Eulate, Julian Theuriet, Christopher J Record, Giorgia Querin, Marion Masingue, Sarah Leonard-Louis, Anthony Behin, Nadine Le Forestier, Antoine Pegat, Maud Michaud, Jean-Baptiste Chanson, Aleksandra Nadaj-Pakleza, Celine Tard, Anne-Laure Bedat-Millet, Guilhem Sole, Marco Spinazzi, Emmanuelle Salort-Campana, Andoni Echaniz-Laguna, Vianney Poinsignon, Philippe Latour, Mary M Reilly, Francoise Bouhour, Tanya Stojkovic
{"title":"Phenotype Presentation and Molecular Diagnostic Yield in Non-5q Spinal Muscular Atrophy.","authors":"Gorka Fernández-Eulate, Julian Theuriet, Christopher J Record, Giorgia Querin, Marion Masingue, Sarah Leonard-Louis, Anthony Behin, Nadine Le Forestier, Antoine Pegat, Maud Michaud, Jean-Baptiste Chanson, Aleksandra Nadaj-Pakleza, Celine Tard, Anne-Laure Bedat-Millet, Guilhem Sole, Marco Spinazzi, Emmanuelle Salort-Campana, Andoni Echaniz-Laguna, Vianney Poinsignon, Philippe Latour, Mary M Reilly, Francoise Bouhour, Tanya Stojkovic","doi":"10.1212/NXG.0000000000200087","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Spinal muscular atrophy (SMA) is mainly caused by homozygous <i>SMN1</i> gene deletions on 5q13. Non-5q SMA patients' series are lacking, and the diagnostic yield of next-generation sequencing (NGS) is largely unknown. The aim of this study was to describe the clinical and genetic landscape of non-5q SMA and evaluate the performance of neuropathy gene panels in these disorders.</p><p><strong>Methods: </strong>Description of patients with non-5q SMA followed in the different neuromuscular reference centers in France as well as in London, United Kingdom. Patients without a genetic diagnosis had undergone at least a neuropathy or large neuromuscular gene panel.</p><p><strong>Results: </strong>Seventy-one patients from 65 different families were included, mostly sporadic cases (60.6%). At presentation, 21 patients (29.6%) showed exclusive proximal weakness (P-SMA), 35 (49.3%) showed associated distal weakness (PD-SMA), and 15 (21.1%) a scapuloperoneal phenotype (SP-SMA). Thirty-two patients (45.1%) had a genetic diagnosis: <i>BICD2</i> (n = 9), <i>DYNC1H1</i> (n = 7), <i>TRPV4</i> (n = 4), <i>VCP</i>, <i>HSBP1</i>, <i>AR</i> (n = 2), <i>VRK1</i>, <i>DNAJB2</i>, <i>MORC2</i>, <i>ASAH1</i>, <i>HEXB</i>, and unexpectedly, <i>COL6A3</i> (n = 1). The genetic diagnostic yield was lowest in P-SMA (6/21, 28.6%) compared with PD-SMA (16/35, 45.7%) and SP-SMA (10/15, 66.7%). An earlier disease onset and a family history of the disease or consanguinity were independent predictors of a positive genetic diagnosis. Neuropathy gene panels were performed in 59 patients with a 32.2% diagnostic yield (19/59). In 13 additional patients, a genetic diagnosis was achieved through individual gene sequencing or an alternative neuromuscular NGS.</p><p><strong>Discussion: </strong>Non-5q SMA is genetically heterogeneous, and neuropathy gene panels achieve a molecular diagnosis in one-third of the patients. The diagnostic yield can be increased by sequencing of other neuromuscular and neurometabolic genes. Nevertheless, there is an unmet need to cluster these patients to aid in the identification of new genes.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 4","pages":"e200087"},"PeriodicalIF":3.7000,"publicationDate":"2023-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3b/83/NXG-2023-000027.PMC10352921.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200087","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objectives: Spinal muscular atrophy (SMA) is mainly caused by homozygous SMN1 gene deletions on 5q13. Non-5q SMA patients' series are lacking, and the diagnostic yield of next-generation sequencing (NGS) is largely unknown. The aim of this study was to describe the clinical and genetic landscape of non-5q SMA and evaluate the performance of neuropathy gene panels in these disorders.
Methods: Description of patients with non-5q SMA followed in the different neuromuscular reference centers in France as well as in London, United Kingdom. Patients without a genetic diagnosis had undergone at least a neuropathy or large neuromuscular gene panel.
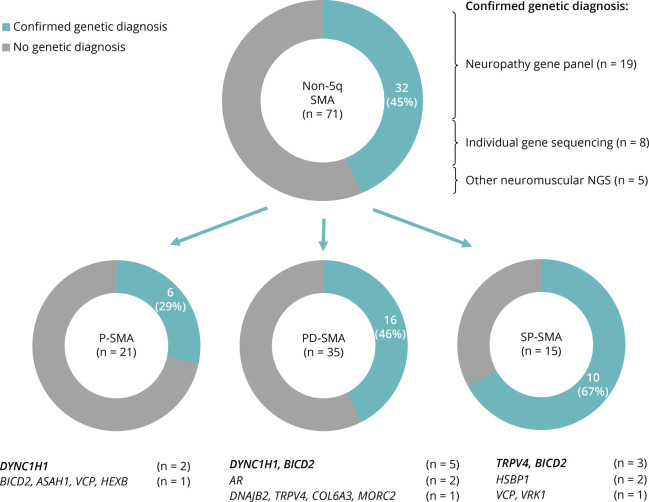
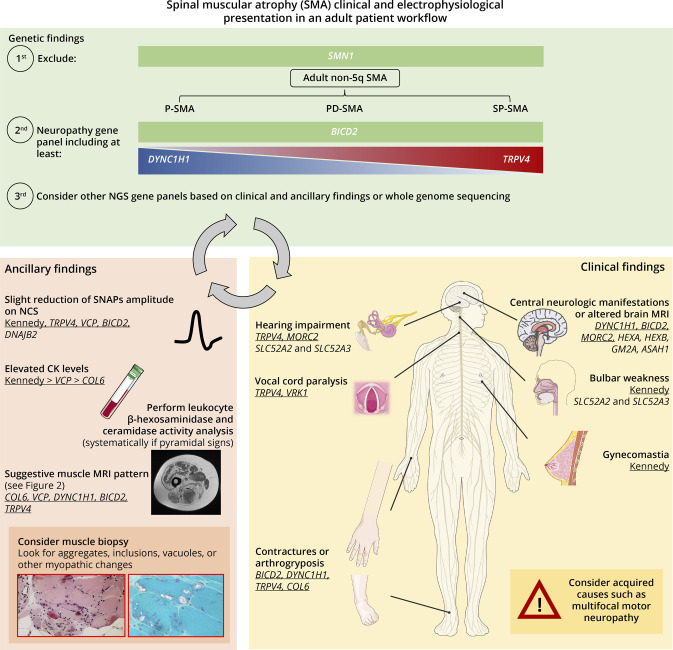
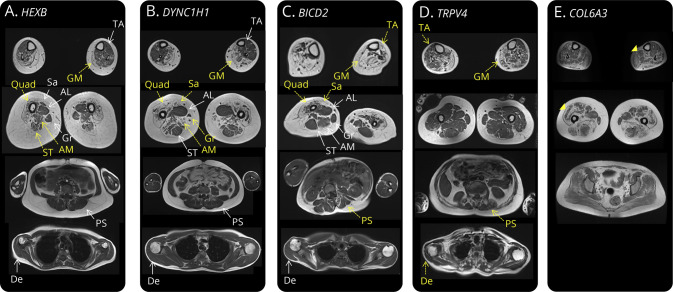
Results: Seventy-one patients from 65 different families were included, mostly sporadic cases (60.6%). At presentation, 21 patients (29.6%) showed exclusive proximal weakness (P-SMA), 35 (49.3%) showed associated distal weakness (PD-SMA), and 15 (21.1%) a scapuloperoneal phenotype (SP-SMA). Thirty-two patients (45.1%) had a genetic diagnosis: BICD2 (n = 9), DYNC1H1 (n = 7), TRPV4 (n = 4), VCP, HSBP1, AR (n = 2), VRK1, DNAJB2, MORC2, ASAH1, HEXB, and unexpectedly, COL6A3 (n = 1). The genetic diagnostic yield was lowest in P-SMA (6/21, 28.6%) compared with PD-SMA (16/35, 45.7%) and SP-SMA (10/15, 66.7%). An earlier disease onset and a family history of the disease or consanguinity were independent predictors of a positive genetic diagnosis. Neuropathy gene panels were performed in 59 patients with a 32.2% diagnostic yield (19/59). In 13 additional patients, a genetic diagnosis was achieved through individual gene sequencing or an alternative neuromuscular NGS.
Discussion: Non-5q SMA is genetically heterogeneous, and neuropathy gene panels achieve a molecular diagnosis in one-third of the patients. The diagnostic yield can be increased by sequencing of other neuromuscular and neurometabolic genes. Nevertheless, there is an unmet need to cluster these patients to aid in the identification of new genes.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
