{"title":"Mutations of SARS-CoV-2 Structural Proteins in the Alpha, Beta, Gamma, and Delta Variants: Bioinformatics Analysis.","authors":"Saima Rehman Khetran, Roma Mustafa","doi":"10.2196/43906","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>COVID-19 and Middle East Respiratory Syndrome are two pandemic respiratory diseases caused by coronavirus species. The novel disease COVID-19 caused by SARS-CoV-2 was first reported in Wuhan, Hubei Province, China, in December 2019, and became a pandemic within 2-3 months, affecting social and economic platforms worldwide. Despite the rapid development of vaccines, there have been obstacles to their distribution, including a lack of fundamental resources, poor immunization, and manual vaccine replication. Several variants of the original Wuhan strain have emerged in the last 3 years, which can pose a further challenge for control and vaccine development.</p><p><strong>Objective: </strong>The aim of this study was to comprehensively analyze mutations in SARS-CoV-2 variants of concern (VoCs) using a bioinformatics approach toward identifying novel mutations that may be helpful in developing new vaccines by targeting these sites.</p><p><strong>Methods: </strong>Reference sequences of the SARS-CoV-2 spike (YP_009724390) and nucleocapsid (YP_009724397) proteins were compared to retrieved sequences of isolates of four VoCs from 14 countries for mutational and evolutionary analyses. Multiple sequence alignment was performed and phylogenetic trees were constructed by the neighbor-joining method with 1000 bootstrap replicates using MEGA (version 6). Mutations in amino acid sequences were analyzed using the MultAlin online tool (version 5.4.1).</p><p><strong>Results: </strong>Among the four VoCs, a total of 143 nonsynonymous mutations and 8 deletions were identified in the spike and nucleocapsid proteins. Multiple sequence alignment and amino acid substitution analysis revealed new mutations, including G72W, M2101I, L139F, 209-211 deletion, G212S, P199L, P67S, I292T, and substitutions with unknown amino acid replacement, reported in Egypt (MW533289), the United Kingdom (MT906649), and other regions. The variants B.1.1.7 (Alpha variant) and B.1.617.2 (Delta variant), characterized by higher transmissibility and lethality, harbored the amino acid substitutions D614G, R203K, and G204R with higher prevalence rates in most sequences. Phylogenetic analysis among the novel SARS-CoV-2 variant proteins and some previously reported β-coronavirus proteins indicated that either the evolutionary clade was weakly supported or not supported at all by the β-coronavirus species.</p><p><strong>Conclusions: </strong>This study could contribute toward gaining a better understanding of the basic nature of SARS-CoV-2 and its four major variants. The numerous novel mutations detected could also provide a better understanding of VoCs and help in identifying suitable mutations for vaccine targets. Moreover, these data offer evidence for new types of mutations in VoCs, which will provide insight into the epidemiology of SARS-CoV-2.</p>","PeriodicalId":73552,"journal":{"name":"JMIR bioinformatics and biotechnology","volume":"4 ","pages":"e43906"},"PeriodicalIF":0.0000,"publicationDate":"2023-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10353769/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMIR bioinformatics and biotechnology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2196/43906","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: COVID-19 and Middle East Respiratory Syndrome are two pandemic respiratory diseases caused by coronavirus species. The novel disease COVID-19 caused by SARS-CoV-2 was first reported in Wuhan, Hubei Province, China, in December 2019, and became a pandemic within 2-3 months, affecting social and economic platforms worldwide. Despite the rapid development of vaccines, there have been obstacles to their distribution, including a lack of fundamental resources, poor immunization, and manual vaccine replication. Several variants of the original Wuhan strain have emerged in the last 3 years, which can pose a further challenge for control and vaccine development.
Objective: The aim of this study was to comprehensively analyze mutations in SARS-CoV-2 variants of concern (VoCs) using a bioinformatics approach toward identifying novel mutations that may be helpful in developing new vaccines by targeting these sites.
Methods: Reference sequences of the SARS-CoV-2 spike (YP_009724390) and nucleocapsid (YP_009724397) proteins were compared to retrieved sequences of isolates of four VoCs from 14 countries for mutational and evolutionary analyses. Multiple sequence alignment was performed and phylogenetic trees were constructed by the neighbor-joining method with 1000 bootstrap replicates using MEGA (version 6). Mutations in amino acid sequences were analyzed using the MultAlin online tool (version 5.4.1).
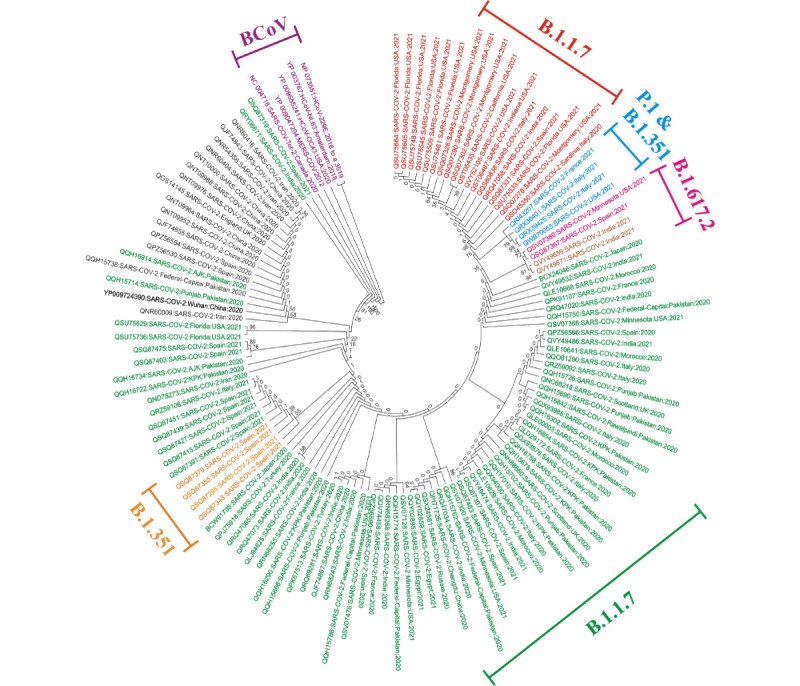
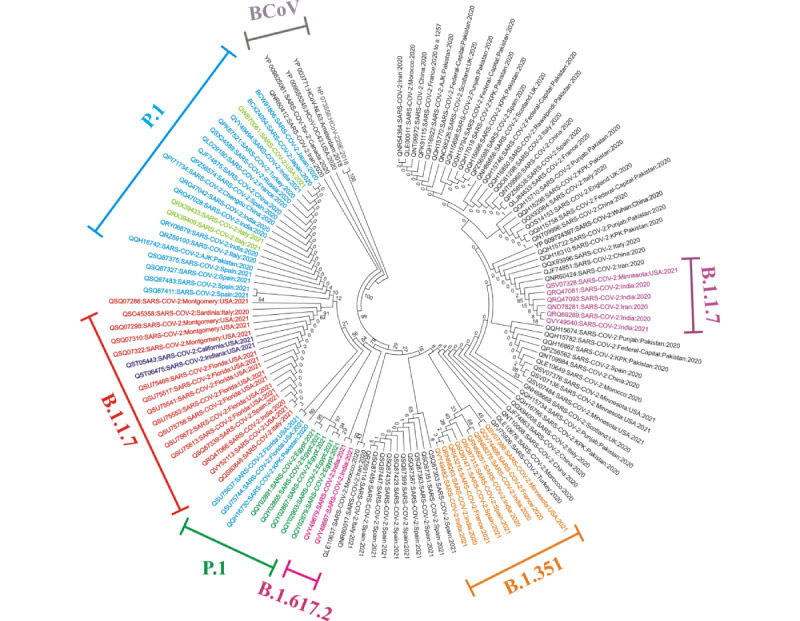
Results: Among the four VoCs, a total of 143 nonsynonymous mutations and 8 deletions were identified in the spike and nucleocapsid proteins. Multiple sequence alignment and amino acid substitution analysis revealed new mutations, including G72W, M2101I, L139F, 209-211 deletion, G212S, P199L, P67S, I292T, and substitutions with unknown amino acid replacement, reported in Egypt (MW533289), the United Kingdom (MT906649), and other regions. The variants B.1.1.7 (Alpha variant) and B.1.617.2 (Delta variant), characterized by higher transmissibility and lethality, harbored the amino acid substitutions D614G, R203K, and G204R with higher prevalence rates in most sequences. Phylogenetic analysis among the novel SARS-CoV-2 variant proteins and some previously reported β-coronavirus proteins indicated that either the evolutionary clade was weakly supported or not supported at all by the β-coronavirus species.
Conclusions: This study could contribute toward gaining a better understanding of the basic nature of SARS-CoV-2 and its four major variants. The numerous novel mutations detected could also provide a better understanding of VoCs and help in identifying suitable mutations for vaccine targets. Moreover, these data offer evidence for new types of mutations in VoCs, which will provide insight into the epidemiology of SARS-CoV-2.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
