{"title":"Primary Hyperoxaluria Type 1: Clinical, Paraclinical, and Evolutionary Aspects in Adults from One Nephrology Center.","authors":"Hajji Meriam, Asma Bettaieb, Hayet Kaaroud, Fethi Ben Hamida, Taher Gargeh, Ridha Mrad, Kahena Bouzid, Ezzeddine Abderrahim","doi":"10.1155/2023/2874414","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Primary hyperoxaluria type 1 (PH1) is a rare and inherited condition of urolithiasis. The aim of our study was to analyze clinical, paraclinical, and evolutionary aspects of PH1 in adult patients in our Nephrology department.</p><p><strong>Methods: </strong>We conducted a retrospective single-center study between 1990 and 2021. We collected patients followed for PH1 confirmed by genetic study and/or histopathological features of renal biopsy and morphoconstitutional analysis of the calculi.</p><p><strong>Results: </strong>There were 25 patients with a gender ratio of 1.78. The median age at onset of symptoms was 18 years. A delay in diagnosis more than 10 years was noted in 13 cases. The genetic study found the I244T mutation in 17 cases and 33-34 InsC in 4 cases. A kidney biopsy was performed in 5 cases, on a native kidney in 4 cases and on a graft biopsy in one case. The analysis of calculi was done in 10 cases showing type Ic in 2 cases. After a median follow-up of 13 years (1 year-42 years), 14 patients progressed to end-stage chronic renal failure (ESRD). The univariate study demonstrated a remarkable association with progression to ESRD in our population (44% vs. 56%) RR = 13.32 (adjusted ORs (95% CI): 2.82-62.79) (<i>p</i> < 0.01).</p><p><strong>Conclusion: </strong>Progression to ESRD was frequent in our series. Early diagnosis and adequate management can delay such an evolution.</p>","PeriodicalId":14177,"journal":{"name":"International Journal of Nephrology","volume":"2023 ","pages":"2874414"},"PeriodicalIF":1.4000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10372328/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Nephrology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/2874414","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 1
Abstract
Introduction: Primary hyperoxaluria type 1 (PH1) is a rare and inherited condition of urolithiasis. The aim of our study was to analyze clinical, paraclinical, and evolutionary aspects of PH1 in adult patients in our Nephrology department.
Methods: We conducted a retrospective single-center study between 1990 and 2021. We collected patients followed for PH1 confirmed by genetic study and/or histopathological features of renal biopsy and morphoconstitutional analysis of the calculi.
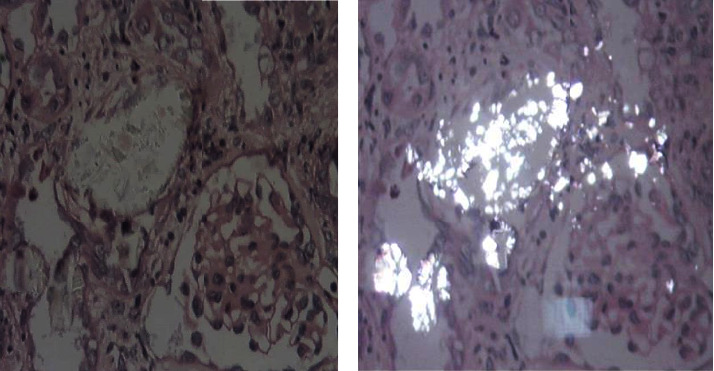
Results: There were 25 patients with a gender ratio of 1.78. The median age at onset of symptoms was 18 years. A delay in diagnosis more than 10 years was noted in 13 cases. The genetic study found the I244T mutation in 17 cases and 33-34 InsC in 4 cases. A kidney biopsy was performed in 5 cases, on a native kidney in 4 cases and on a graft biopsy in one case. The analysis of calculi was done in 10 cases showing type Ic in 2 cases. After a median follow-up of 13 years (1 year-42 years), 14 patients progressed to end-stage chronic renal failure (ESRD). The univariate study demonstrated a remarkable association with progression to ESRD in our population (44% vs. 56%) RR = 13.32 (adjusted ORs (95% CI): 2.82-62.79) (p < 0.01).
Conclusion: Progression to ESRD was frequent in our series. Early diagnosis and adequate management can delay such an evolution.
期刊介绍:
International Journal of Nephrology is a peer-reviewed, Open Access journal that publishes original research articles, review articles, and clinical studies focusing on the prevention, diagnosis, and management of kidney diseases and associated disorders. The journal welcomes submissions related to cell biology, developmental biology, genetics, immunology, pathology, pathophysiology of renal disease and progression, clinical nephrology, dialysis, and transplantation.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
