Amir Hossein Darooneh, Michelle Przedborski, Mohammad Kohandel
{"title":"A novel statistical method predicts mutability of the genomic segments of the SARS-CoV-2 virus.","authors":"Amir Hossein Darooneh, Michelle Przedborski, Mohammad Kohandel","doi":"10.1017/qrd.2021.13","DOIUrl":null,"url":null,"abstract":"<p><p>The SARS-CoV-2 virus has made the largest pandemic of the 21st century, with hundreds of millions of cases and tens of millions of fatalities. Scientists all around the world are racing to develop vaccines and new pharmaceuticals to overcome the pandemic and offer effective treatments for COVID-19 disease. Consequently, there is an essential need to better understand how the pathogenesis of SARS-CoV-2 is affected by viral mutations and to determine the conserved segments in the viral genome that can serve as stable targets for novel therapeutics. Here, we introduce a text-mining method to estimate the mutability of genomic segments directly from a reference (ancestral) whole genome sequence. The method relies on calculating the importance of genomic segments based on their spatial distribution and frequency over the whole genome. To validate our approach, we perform a large-scale analysis of the viral mutations in nearly 80,000 publicly available SARS-CoV-2 predecessor whole genome sequences and show that these results are highly correlated with the segments predicted by the statistical method used for keyword detection. Importantly, these correlations are found to hold at the codon and gene levels, as well as for gene coding regions. Using the text-mining method, we further identify codon sequences that are potential candidates for siRNA-based antiviral drugs. Significantly, one of the candidates identified in this work corresponds to the first seven codons of an epitope of the spike glycoprotein, which is the only SARS-CoV-2 immunogenic peptide without a match to a human protein.</p>","PeriodicalId":34636,"journal":{"name":"QRB Discovery","volume":"3 ","pages":"e1"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8795775/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"QRB Discovery","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1017/qrd.2021.13","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 1
Abstract
The SARS-CoV-2 virus has made the largest pandemic of the 21st century, with hundreds of millions of cases and tens of millions of fatalities. Scientists all around the world are racing to develop vaccines and new pharmaceuticals to overcome the pandemic and offer effective treatments for COVID-19 disease. Consequently, there is an essential need to better understand how the pathogenesis of SARS-CoV-2 is affected by viral mutations and to determine the conserved segments in the viral genome that can serve as stable targets for novel therapeutics. Here, we introduce a text-mining method to estimate the mutability of genomic segments directly from a reference (ancestral) whole genome sequence. The method relies on calculating the importance of genomic segments based on their spatial distribution and frequency over the whole genome. To validate our approach, we perform a large-scale analysis of the viral mutations in nearly 80,000 publicly available SARS-CoV-2 predecessor whole genome sequences and show that these results are highly correlated with the segments predicted by the statistical method used for keyword detection. Importantly, these correlations are found to hold at the codon and gene levels, as well as for gene coding regions. Using the text-mining method, we further identify codon sequences that are potential candidates for siRNA-based antiviral drugs. Significantly, one of the candidates identified in this work corresponds to the first seven codons of an epitope of the spike glycoprotein, which is the only SARS-CoV-2 immunogenic peptide without a match to a human protein.
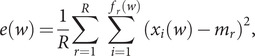
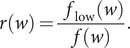
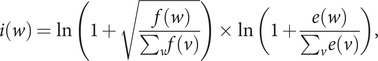



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
