{"title":"Ocular Manifestations of Hurler-Scheie Syndrome: Recurrence of Host Disease in the Corneal Transplant.","authors":"Zsófia Kölkedi, Adrienne Csutak, Eszter Szalai","doi":"10.1159/000525453","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Hurler-Scheie syndrome is a type of mucopolysaccharidosis I (MPS). In MPS I the decreased activity of alpha-L-iduronidase lysosomal enzyme leads to glycosaminoglycan (GAG) deposition in the intra- and extracellular matrix. Excessive amounts of GAG can accumulate in most layers of the cornea, including epithelial cells, stromal keratocytes, and endothelial cells.</p><p><strong>Case presentation: </strong>A 25-year-old female patient suffering from Hurler-Scheie syndrome with multiple ocular manifestations is reported. Due to significant bilateral corneal opacification, penetrating keratoplasty was performed on both eyes. Histopathologic examination of the corneal buttons showed disorganized collagen fibers with heterogenous thickness and many granule-containing keratocytes with excessive cytoplasm. Despite receiving enzyme replacement therapy, in vivo confocal microscopy revealed characteristic vacuoles in the basal epithelium and corneal stroma 96 months after transplantation. High resolution anterior segment optical coherence tomography demonstrated hyperreflective opacities superficial and deeper in the stroma which was consistent with recurrence of host disease in the graft.</p><p><strong>Conclusion: </strong>To the best of our knowledge, this is the first documented Hurler-Scheie syndrome case of recurrence after penetrating keratoplasty demonstrated by in vivo confocal microscopy. Additionally, this patient manifested severe ocular involvement of MPS which might be an explanation of the progressive course of corneal opacification after transplantation.</p>","PeriodicalId":48566,"journal":{"name":"Molecular Syndromology","volume":"14 1","pages":"44-50"},"PeriodicalIF":0.9000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/30/38/msy-0014-0044.PMC9911992.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Syndromology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000525453","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Hurler-Scheie syndrome is a type of mucopolysaccharidosis I (MPS). In MPS I the decreased activity of alpha-L-iduronidase lysosomal enzyme leads to glycosaminoglycan (GAG) deposition in the intra- and extracellular matrix. Excessive amounts of GAG can accumulate in most layers of the cornea, including epithelial cells, stromal keratocytes, and endothelial cells.
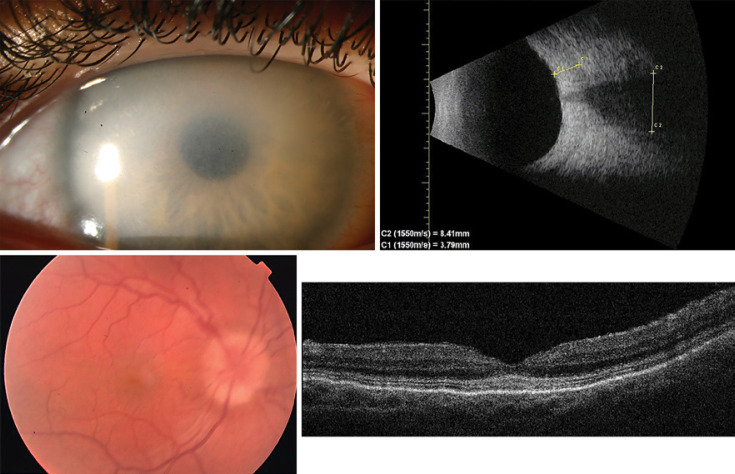
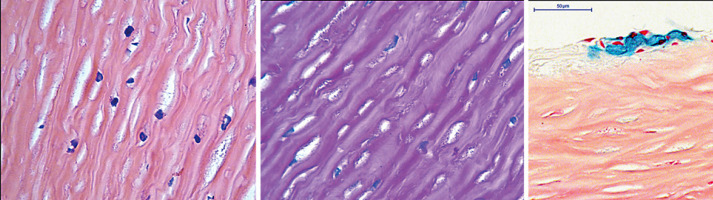
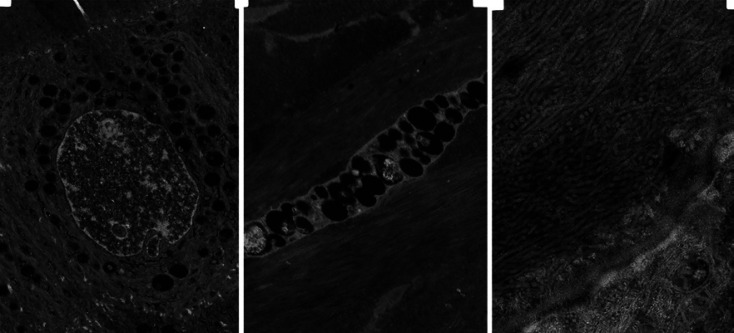
Case presentation: A 25-year-old female patient suffering from Hurler-Scheie syndrome with multiple ocular manifestations is reported. Due to significant bilateral corneal opacification, penetrating keratoplasty was performed on both eyes. Histopathologic examination of the corneal buttons showed disorganized collagen fibers with heterogenous thickness and many granule-containing keratocytes with excessive cytoplasm. Despite receiving enzyme replacement therapy, in vivo confocal microscopy revealed characteristic vacuoles in the basal epithelium and corneal stroma 96 months after transplantation. High resolution anterior segment optical coherence tomography demonstrated hyperreflective opacities superficial and deeper in the stroma which was consistent with recurrence of host disease in the graft.
Conclusion: To the best of our knowledge, this is the first documented Hurler-Scheie syndrome case of recurrence after penetrating keratoplasty demonstrated by in vivo confocal microscopy. Additionally, this patient manifested severe ocular involvement of MPS which might be an explanation of the progressive course of corneal opacification after transplantation.
简介:Hurler-Scheie综合征是粘多糖病(MPS)的一种。在MPS I中,α - l -伊杜糖醛酸酶溶酶体酶活性降低导致糖胺聚糖(GAG)沉积在细胞内和细胞外基质中。过量的GAG可积聚在角膜的大多数层,包括上皮细胞、间质角质细胞和内皮细胞。病例介绍:我们报告了一位25岁的女性患者患有多发性眼部表现的赫勒-谢氏综合征。由于严重的双侧角膜混浊,我们对双眼进行了穿透性角膜移植术。角膜钮扣的组织病理学检查显示胶原纤维紊乱,厚度不均,有许多含颗粒的角质细胞,细胞质过多。尽管接受了酶替代治疗,活体共聚焦显微镜在移植后96个月发现基底上皮和角膜基质中有特征性的空泡。高分辨率前段光学相干断层扫描显示基质表面和深层的高反射性混浊,这与移植物中宿主疾病的复发一致。结论:据我们所知,这是首例通过体内共聚焦显微镜观察穿透性角膜移植术后复发的赫勒-谢氏综合征病例。此外,该患者表现出严重的MPS眼部受累,这可能解释了移植后角膜混浊的进行性过程。
期刊介绍:
''Molecular Syndromology'' publishes high-quality research articles, short reports and reviews on common and rare genetic syndromes, aiming to increase clinical understanding through molecular insights. Topics of particular interest are the molecular basis of genetic syndromes, genotype-phenotype correlation, natural history, strategies in disease management and novel therapeutic approaches based on molecular findings. Research on model systems is also welcome, especially when it is obviously relevant to human genetics. With high-quality reviews on current topics the journal aims to facilitate translation of research findings to a clinical setting while also stimulating further research on clinically relevant questions. The journal targets not only medical geneticists and basic biomedical researchers, but also clinicians dealing with genetic syndromes. With four Associate Editors from three continents and a broad international Editorial Board the journal welcomes submissions covering the latest research from around the world.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
