{"title":"Autosomal Recessive Spinocerebellar Ataxia Type 9 With a Response to Phosphate Repletion: A Case Report.","authors":"Shotaro Haji, Ryosuke Miyamoto, Hiroyuki Morino, Yusuke Osaki, Seijiro Tsuji, Ichizo Nishino, Masahiro Abe, Yuishin Izumi","doi":"10.1212/NXG.0000000000200070","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Autosomal recessive spinocerebellar ataxia type 9 (SCAR9) has received attention due to its potential response to coenzyme Q10 (CoQ10) supplementation; however, the response has so far been limited and variable.</p><p><strong>Methods: </strong>We report a SCAR9 patient with severe hypophosphatemia who responded well to CoQ10 and phosphate repletion.</p><p><strong>Results: </strong>A 70-year-old man (the offspring of a consanguineous marriage) presented with cerebellar ataxia and intense fatigue after exercise. Whole-exome sequencing identified a novel homozygous deletion mutation (NM_020247.5:c.1218_1219del) in <i>COQ8A</i>. We thus diagnosed him with SCAR9. Supplementation of CoQ10 alleviated his symptoms, with the Scale for the Assessment and Rating of Ataxia (SARA) dropping from 16 to 14. During the course of the disease, he demonstrated continuous hypophosphatemia caused by renal phosphate wasting. Gait dysfunction due to weakness and eye movement was partially alleviated, and SARA dropped from 17 to 13 after phosphate repletion.</p><p><strong>Discussion: </strong>Phosphate repletion should be considered for patients with severe hypophosphatemia without any apparent subjective symptoms. In this case, phosphate repletion could have improved myopathy leading to partial improvement in the patient's symptoms. Further analyses regarding the association between COQ8A mutation and phosphate wasting are required to elucidate the detailed pathogenesis.</p><p><strong>Classification of evidence: </strong>This provides Class IV evidence. This is a single observational study without controls.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"9 3","pages":"e200070"},"PeriodicalIF":3.7000,"publicationDate":"2023-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/85/96/NXG-2023-000013.PMC10389171.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200070","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Objective: Autosomal recessive spinocerebellar ataxia type 9 (SCAR9) has received attention due to its potential response to coenzyme Q10 (CoQ10) supplementation; however, the response has so far been limited and variable.
Methods: We report a SCAR9 patient with severe hypophosphatemia who responded well to CoQ10 and phosphate repletion.
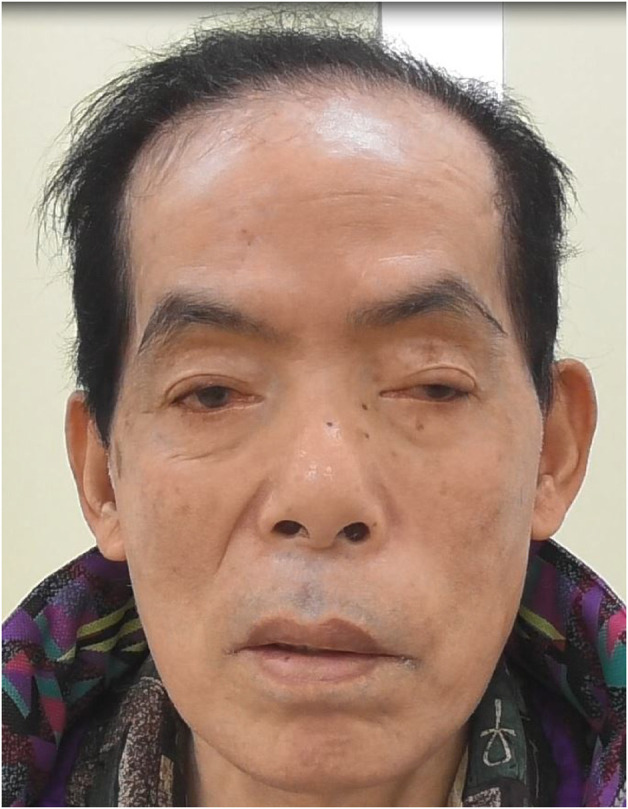
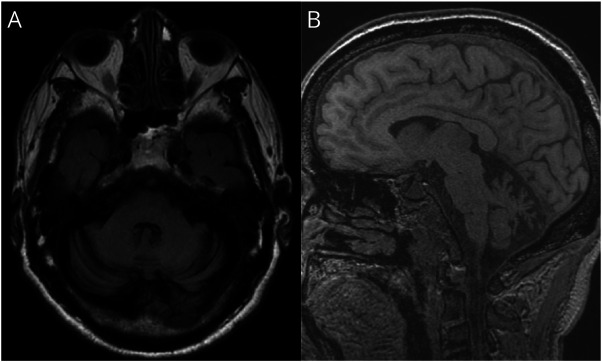
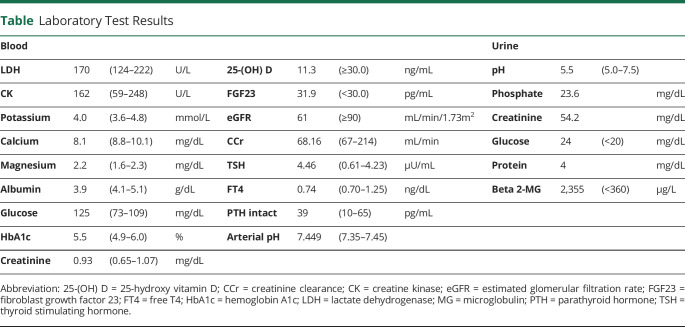
Results: A 70-year-old man (the offspring of a consanguineous marriage) presented with cerebellar ataxia and intense fatigue after exercise. Whole-exome sequencing identified a novel homozygous deletion mutation (NM_020247.5:c.1218_1219del) in COQ8A. We thus diagnosed him with SCAR9. Supplementation of CoQ10 alleviated his symptoms, with the Scale for the Assessment and Rating of Ataxia (SARA) dropping from 16 to 14. During the course of the disease, he demonstrated continuous hypophosphatemia caused by renal phosphate wasting. Gait dysfunction due to weakness and eye movement was partially alleviated, and SARA dropped from 17 to 13 after phosphate repletion.
Discussion: Phosphate repletion should be considered for patients with severe hypophosphatemia without any apparent subjective symptoms. In this case, phosphate repletion could have improved myopathy leading to partial improvement in the patient's symptoms. Further analyses regarding the association between COQ8A mutation and phosphate wasting are required to elucidate the detailed pathogenesis.
Classification of evidence: This provides Class IV evidence. This is a single observational study without controls.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
